Primary Immunodeficiency
Primary Immunodeficiency Pathology Video
Primary immune deficiency diseases (PIDDs) are uncommon immune system-damaging hereditary illnesses.
Primary immune deficiency illnesses come in more than 200 different varieties.
A few examples of primary immunodeficiency includes:
- Congenital neutropenia syndrome
- Autoimmune lymphoproliferative syndrome (ALPS)
- Autoimmune polyglandular syndrome type 1
- BENTA disease
- Caspase 8 deficiency syndrome (CEDS)
- Chronic granulomatous disease
- DiGeorge syndrome
- Severe combined immunodeficiency (SCID)
- X linked agammaglobulinemia
- Common variable immunodeficiency (CVID)
- IgA deficiency
- Hyper-IgM syndrome
- Complement deficiencies
Low amounts of neutrophils are a hallmark of the congenital neutropenia syndromes.
The rare immunological condition known as autoimmune lymphoproliferative syndrome can lead to a variety of autoimmune problems.
Autoimmune polyglandular syndrome type 1 is characterized by a wide range of symptoms, including autoimmunity to various organs and an increased susceptibility to candidiasis (a fungal infection caused by Candida yeast).
BENTA disease is a rare genetic immune system disorder caused by mutations in the CARD11 gene.
Caspase 8 deficiency state is a very rare immune system genetic disorder caused by mutations in the CASP8 gene.
Chronic granulomatous disease (CGD) occurs when white blood cells known as phagocytes are unable to kill specific bacteria and fungi, leaving people vulnerable to bacterial and fungal infections.
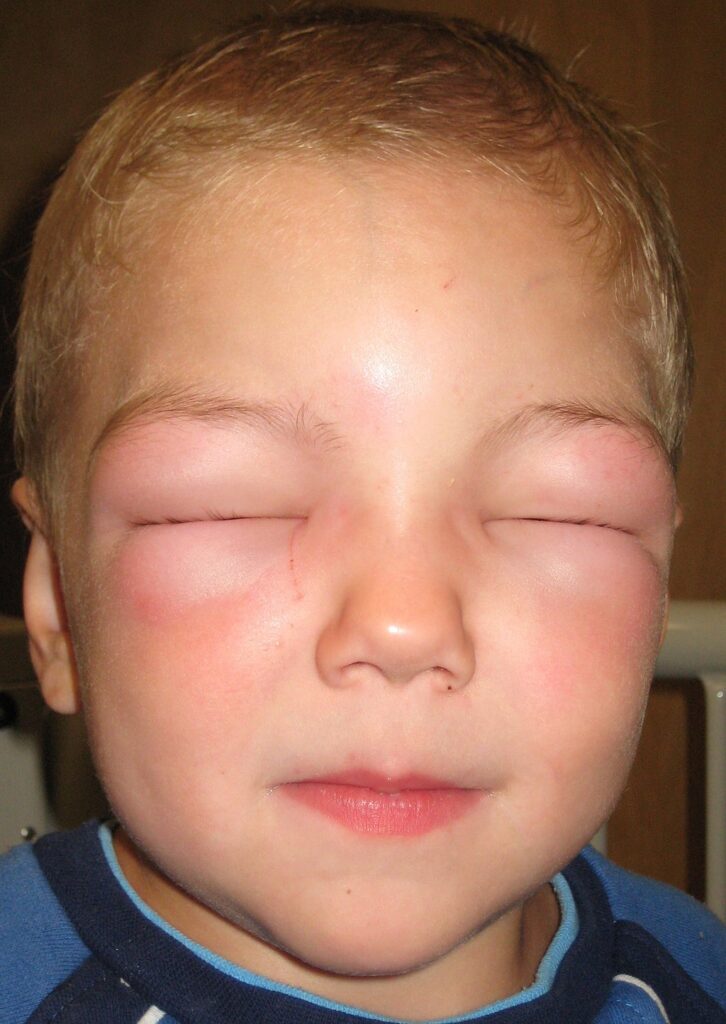
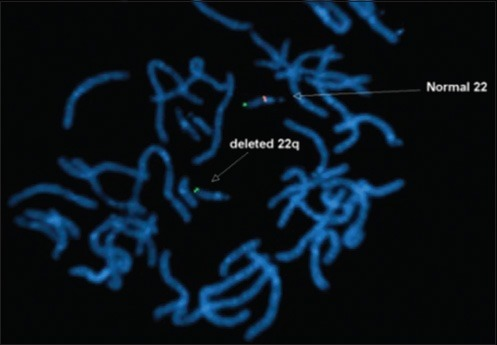
DiGeorge Syndrome
DiGeorge syndrome is a congenital condition due to 22q11 microdeletion.
The condition’s severity varies.
DiGeorge syndrome results from failure of the third and fourth pharyngeal pouches to develop.
The symptoms of DiGeorge syndrome include:
- Hypocalcemia
- Infections brought on by a T-cell shortage brought on by the absence of a thymus
- Dysmorphic facies
- Heart defects
- Learning issues
Severe Combined Immunodeficiency (SCID)
Severe combined immunodeficiency (SCID) may be caused by either:
- Deficiencies in adenosine deaminase (ADA)
- Defects in cytokine receptors
- MHC class II abnormalities which predisposes to dysfunctional CD4+ T cells
Deficiencies in adenosine deaminase (ADA)
Adenosine and deoxyadenosine must be deaminated in order to be excreted as waste products due to adenosine deaminase (ADA) insufficiency.
Defects in cytokine receptors
Cytokine signaling is required for the proliferation and maturation of B and T cells, and cytokine receptor abnormalities are among the etiologies.
MHC class II abnormalities which predisposes to dysfunctional CD4+ T cells
MHC class II is required for CD4+ helper T cell activation and cytokine generation.
SCID is associated with defective cell-mediated and humoral immunity.
SCID is characterized by recurrent infections with a variety of organisms, including live vaccines, opportunistic infections, fungal, viruses, bacteria, and protozoa.
SCID is treated by:
- Sterile isolation
- Stem cell transplant
Lymphocytes are toxic when adenosine and deoxyadenosine accumulate.
There is increased susceptibility to bacterial, viral, fungal, and protozoal infections, as well as opportunistic infections and live vaccinations, characterizes this condition.
Stem cell transplantation and sterile isolation, also known as bubble babies, are treatments.

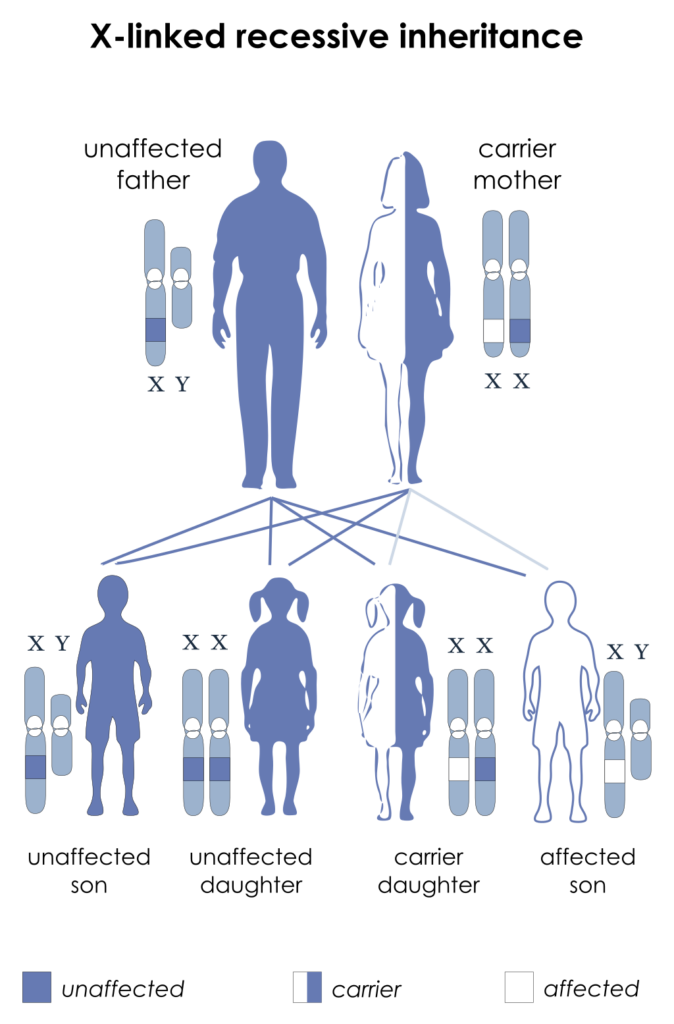
X-Linked Agammaglobulinemia
X linked agammaglobulinemia is a hereditary (genetic) immune system condition that makes it harder to fight infections.
X linked agammaglobulinemia is characterized as complete lack of immunoglobulin due to disorderedness.
B-cell maturation is impacted, and naive B cells do not mature to plasma cells.
X linked agammaglobulinemia is due to mutated Bruton tyrosine kinase.
X linked agammaglobulinemia is an X-linked disease.
During the first six months of life, maternal antibodies are present and protective.
After approximately six months of life, this syndrome manifests as recurring bacterial, enterovirus (such coxsackievirus and poliovirus), and Giardia Lamblia infections.
Patients with X linked agammaglobulinemia should avoid Polio vaccinations and other live vaccines.
Common Variable Immunodeficiency (CVID)
A broad collection of diseases known as common variable immunodeficiency (CVID) are characterized by hypogammaglobulinemia and recurrent infections that are primarily bacterial.
Defects in the helper T-cells or the B-cells cause low immunoglobulin levels.
Bacterial, enterovirus, and Giardia infections are more likely to occur, mainly in late childhood.
Increased risk for autoimmune disease and lymphoma.

IgA Deficiency
IgA deficiency is characterized by decreased mucosal and serum lgA.
IgA deficiency is the most common immunoglobulin deficiency.
IgA deficiency is common in patients with celiac disease.
Most patients with IgA deficiency typically asymptomatic, but many are prone to mucosal infections, particularly viral infections.
Hyper-IgM Syndrome
Hyper-IgM syndrome is associated with elevated levels of lgM.
Hyper-IgM syndrome is caused by mutant CD40L on helper T cells or CD40 receptor on B cells.
Helper T cells cannot receive a second signal when B-cells are activated.
The blocked signal does not allow the cytokines required for isotype class switching.
Immunoglobulin classes (IgA, IgG, IgE) are not produced.
Low IgA, IgG, and lgE levels result in poor opsonization of pathogens, especially at mucosal locations.
Hyper-IgM syndrome is characterized by recurrent pyogenic infections.
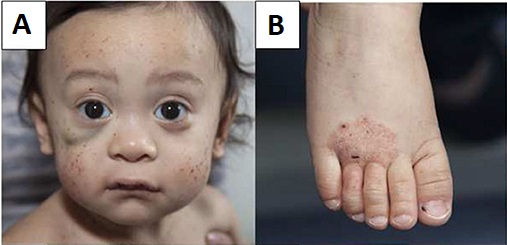
Wiskott-Aldrich Syndrome
Wiskott-Aldrich syndrome is due to defective humoral and cellular immunity.
Wiskott-Aldrich syndrome is caused by mutation an X-linked mutation in the Wiskott-Aldrich Syndrome protein (WASP) gene.
Wiskott-Aldrich syndrome features include:
- Thrombocytopenia
- Eczema
- Recurrent infections
Complement Deficiencies
Patients with complement deficiencies (C5-C9 deficiencies) have recurrent Neisseria species infections in their systems (N. meningitidis and N. gonorrhea).
Meningococcal infections with unusual serotypes (W-135, X, Y, and Z) are more common than those with common serotypes (A, B, and C).
Hereditary angioedema, which is characterized by edema of the skin, especially periorbital and mucosal regions, is caused by a deficit in the C1 inhibitor.