Primary Hemostasis
Primary Hemostasis Pathology Video
Step 1: Transient Vasoconstriction of Damaged Vessel
A vascular spasm is activated which leads to vasoconstriction.
The vascular spasm is mediated by:
- Neural reflex
- Contraction of endothelial cells due to endothelin being released from neighboring endothelial cells that were damaged
Step 2: Platelet Adhesion to the Surface of Disrupted Vessel
Platelet adhesion takes place as a result of interaction between various inflammatory mediators released by the extracellular matrix (ECM).
One of the main mediators of platelet adhesion is the von Willebrand factor (vWF).
Von Willebrand factor binds to the GPIb receptor and results in the attachment of the platelet to the damaged vessel.
Von Willebrand factor is derived from:
- Weibel-Palade bodies of endothelial cells
- Alpha-granules of platelets
Step 3: Platelet Degranulation/Activation
Adhesion of platelets results in a change in platelet conformation.
Platelets release cytoplasmic granules which contain factors such as:
- ADP
- Thromboxane A2 (TXA2)
Platelets also undergo a change in shape and become pseudopodal under the influence of P2Y1 receptors.
This leads to the release of various chemokines.
Step 4: Platelet Aggregation
As a consequence of platelet aggregation, platelets form an aggregation called a platelet plug.
This platelet plug is weak.
The platelet plug is further stabilized by the coagulation cascade.
The coagulation cascade is part of secondary hemostasis.
 Primary Hemostasis. Modern coagulation pathway. Hand-drawn composite from similar drawings presented by Professor Dzung Le, MD, PhD, at UCSD Clinical Chemistry conferences on 14 and 21 October 2014. Original schema from Introduction to Hematology by Samuel I. Rapaport. 2nd edition;Lippencott:1987. Dr Le added the factor XI portion based on a paper from about year 2000. Dr. Le's similar drawings presented the development of this cascade over 6 frames, like a comic. Niels Olson . Not altered. CC BY-SA 4.0
Primary Hemostasis. Modern coagulation pathway. Hand-drawn composite from similar drawings presented by Professor Dzung Le, MD, PhD, at UCSD Clinical Chemistry conferences on 14 and 21 October 2014. Original schema from Introduction to Hematology by Samuel I. Rapaport. 2nd edition;Lippencott:1987. Dr Le added the factor XI portion based on a paper from about year 2000. Dr. Le's similar drawings presented the development of this cascade over 6 frames, like a comic. Niels Olson . Not altered. CC BY-SA 4.0


Disorders of Primary Hemostasis
Disorders of primary homeostasis are typically due to abnormalities in platelets.
Disorders of primary hemostasis are divided into:
- Quantitative platelet disorders
- Qualitative platelet disorders
Clinical features of primary hemostasis include:
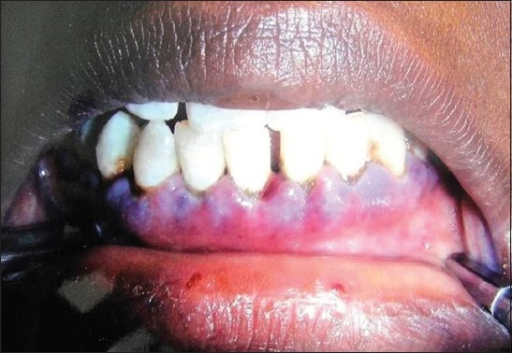
- Mucosal bleeding
- Skin bleeding
- Ecchymoses
- Petechiae
- Hematomas
- Prolonged bleeding after minor surgeries
Mucosal bleeding presents as:
- Epistaxis
- Gingival bleeding
- Hematuria
- Hemoptysis
- Hematemesis
- Menorrhagia
Severe thrombocytopenia may result in intracranial bleeding.
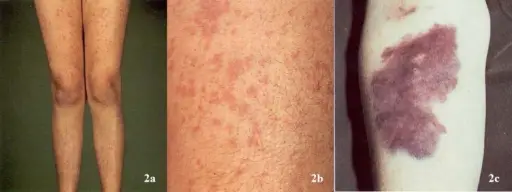
Symptoms of skin bleeding include:
- Petechiae (1-2 mm)
- Ecchymoses (> 1 cm)
- Purpura (> 3 mm)
- Easy bruising
Petechiae are a sign of thrombocytopenia and are not usually seen with qualitative disorders.
Useful laboratory studies include:
- Bleeding time (BT)
- Platelet count
- Blood smear
- Bone marrow biopsy (BMBx)
Bleeding time (BT) is normally 2-7 minutes.
In primary hemostatic disorders bleeding time (BT) is prolonged.
Platelet count is normally 150,000 – 450,000 per microliter.
Blood smears are used to assess number and size of platelets.
Bone marrow biopsy is used to assess megakaryocytes, which produce platelets.
Spontaneous bleeding can occur with a platelet count under 10,000/microliter and surgical bleeding with counts below 50,000/microliter.
 In vivo cartoon representation of the multiple scales at which haemostasis occurs. Biomechanics of haemostasis and thrombosis in health and disease: from the macro- to molecular scale. Tran R, Myers DR, Ciciliano J, Trybus Hardy EL, Sakurai Y, Ahn B, Qiu Y, Mannino RG, Fay ME, Lam WA - Journal of cellular and molecular medicine (2013). Not Altered. CC.
In vivo cartoon representation of the multiple scales at which haemostasis occurs. Biomechanics of haemostasis and thrombosis in health and disease: from the macro- to molecular scale. Tran R, Myers DR, Ciciliano J, Trybus Hardy EL, Sakurai Y, Ahn B, Qiu Y, Mannino RG, Fay ME, Lam WA - Journal of cellular and molecular medicine (2013). Not Altered. CC.
Immune Thrombocytopenia Purpura (ITP)
Immune thrombocytopenia purpura (ITP) is an autoimmune disorder characterized by the formation of IgG antibodies against glycoprotein IIb/IIIa (GPIIb/IIIa) receptors and their destruction.
Immune thrombocytopenia purpura (ITP) is the most common cause of thrombocytopenia in children and adults.
Immune thrombocytopenia purpura (ITP) is an isolated thrombocytopenia.
Hematocrit and white blood cell (WBC) count is normal in Immune thrombocytopenia purpura (ITP).
Autoantibodies are produced by plasma cells in the spleen.
Antibody-bound platelets are consumed by splenic macrophages, resulting in thrombocytopenia.
Immune thrombocytopenia purpura (ITP) is divided into acute and chronic forms.
Acute immune thrombocytopenia purpura (ITP):
- Arises in children weeks after a viral infection or immunization
- Usually resolves within weeks of presentation
Chronic immune thrombocytopenia purpura (ITP):
- Arises in adults
- Usually women of child-bearing age
- May be primary
- May be secondary (e.g. related to systemic lupus erythematous (SLE)
Note that immune thrombocytopenia purpura (ITP) in a new mother may cause short-lived thrombocytopenia in a newborn offspring because antiplatelet IgG can cross the placenta.
Laboratory findings of Immune thrombocytopenia purpura (ITP) include:
- Platelet count < 50
- Normal PT/PTT
- Normal coagulation factors
- Megakaryocytes in bone marrow biopsy
Initial treatment of Immune thrombocytopenia purpura (ITP) is corticosteroids:
- Children typically respond well to corticosteroids
- Adults may show early response to corticosteroids, but often relapse
IVIg is used to raise the platelet count in symptomatic bleeding, but its effect is short.
In refractory cases, a splenectomy may be performed to eliminate the primary source of antibodies and the site of platelet destruction (performed in refractory cases).
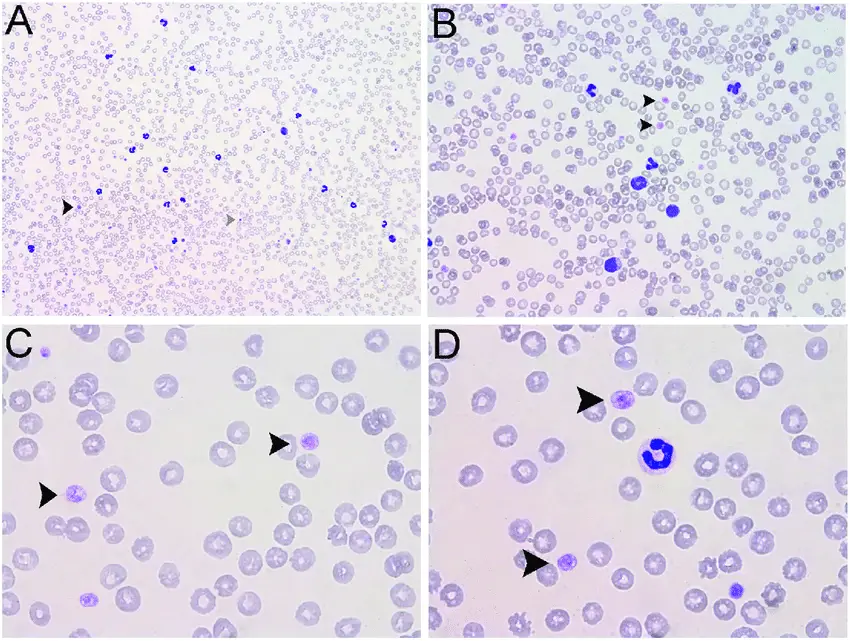
 Immune Thrombocytopenia Purpura. Blood film showing giant platelets - arrows - in a person with ITP (Giemsa stain) Dr Graham Beards - Not altered. CC BY-SA 4.0
Immune Thrombocytopenia Purpura. Blood film showing giant platelets - arrows - in a person with ITP (Giemsa stain) Dr Graham Beards - Not altered. CC BY-SA 4.0





Microangiopathic Hemolytic Anemia
Microangiopathic hemolytic anemia (MAHA) is the pathologic formation of platelet microthrombi in small vessels.
Microangiopathic hemolytic anemia means small blood vessels with pathology and hemolytic anemia.
Platelets are used in the formation of microthrombi.
Red blood cells (RBCs) are sheared as they interact with the microthrombi, resulting in hemolytic anemia with schistocytes.
Microangiopathic hemolytic anemia is seen in thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS).
Thrombotic Thrombocytopenic Purpura (TTP)
TTP is due to decreased ADAMTS13.
ADAMTS13 is an enzyme that normally cleaves von Willebrand factor (vWF) multimers into smaller monomers for eventual degradation.
Decreased ADAMTS13 is usually due to an acquired autoantibody, and is most commonly seen in adult females.
The large uncleaved vWF multimers of TTP lead to abnormal platelet adhesion, that result in microthrombi formation.
Hemolytic Uremic Syndrome (HUS)
Hemolytic uremic syndrome (HUS) is due to endothelial damage by drugs or infection.
HUS is classically seen in children with E. coli O157:H7 dysentery.
Exposure to undercooked beef is a risk factor for this condition.
E. coli verotoxin damages endothelial cells resulting in platelet microthrombi.
Clinical findings of HUS and TTP include:
- Skin bleeding
- Mucosal bleeding
- Microangiopathic hemolytic anemia
- Fever
- Renal insufficiency (more common in HUS, thrombi involve vessels of the kidney)
- CNS abnormalities (more common in TTP, thrombi involve vessels of the CNS)
Laboratory findings of HUS and TTP include:
- Thrombocytopenia
- Increased bleeding time (BT)
- Normal PT/ PTT
- Normal coagulation cascade and factors
- Amenia
- Schistocytes
- Megakaryocytes in the bone marrow biopsy
Treatment of HUS and TTP involves plasmapheresis and corticosteroids, particularly in TTP.
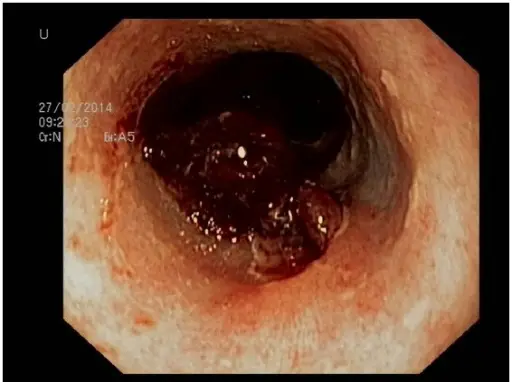
 Microangiopathic Hemolytic Anemia. DIC With Microangiopathic Hemolytic Anemia 34 y/o female, Hb 8.6 g/dL, MCV 104.5 fL, MCHC 32.8 g/dL, platelets 11,000/uL, WBC 59,000/uL. Patient had a history of disseminated non-small cell carcinoma of the lung. She presented to the ER in extremis and expired within a few hours of admission. Morphology: Thrombocytopenia, 4+ schizocytes, 3+ spherocytes, 4+ polychromatophilic rbc. Diagnosis: Disseminated carcinomatosis with DIC. Ed Uthman, MD. Not altered. Public Domain
Microangiopathic Hemolytic Anemia. DIC With Microangiopathic Hemolytic Anemia 34 y/o female, Hb 8.6 g/dL, MCV 104.5 fL, MCHC 32.8 g/dL, platelets 11,000/uL, WBC 59,000/uL. Patient had a history of disseminated non-small cell carcinoma of the lung. She presented to the ER in extremis and expired within a few hours of admission. Morphology: Thrombocytopenia, 4+ schizocytes, 3+ spherocytes, 4+ polychromatophilic rbc. Diagnosis: Disseminated carcinomatosis with DIC. Ed Uthman, MD. Not altered. Public Domain

Qualitative Platelet Disorders
Qualitative platelet disorders are due to low quality platelets.
In qualitative platelet disorders:
- The number of platelets present may be normal
- The structure and or function of the platelets is low quality
Examples of qualitative platelet disorders include:
- Bernard-Soulier syndrome
- Glanzmann thrombasthenia
Bernard-Soulier Syndrome
Bernard-Soulier syndrome is due to a genetic glycoprotein Ib deficiency which impairs platelet adhesion.
Blood smear shows mild thrombocytopenia with enlarged platelets.
Glanzmann Thrombasthenia
Glanzmann thrombasthenia is due to a genetic GPIb/Ia deficiency which impairs platelet aggregation.
Aspirin irreversibly inactivates cyclooxygenase (COX).
Lack of thromboxane A2 (TXA2) impairs platelet aggregation.
Uremia also disrupts platelet function, both adhesion and aggregation are impaired.