Genetic disorders are conditions that are caused by an abnormality in an individual’s DNA.
What Are Mutations?
Mutations are alterations in the nucleotide sequence of the genome of an organism, virus, or extrachromosomal DNA.
Examples of mutations include:
- Point mutations
- Mutations in noncoding sequences
- Mutations in introns
- Mutations in exons
- Deletions
- Insertions
- Trinucleotide repeats
| Point mutations | A point mutation is alteration of a single base pair. The base substitution can be a silent mutation, which produces the same amino acid. The base substitution can be a missense mutation where the altered codon produces a different amino acid. The base substitution can be a nonsense mutation where a stop signal is produced. | – Point mutations are present in multiple tumor suppressor proteins cause cancer. Adenomatous Polyposis Col. Neurofibromatosis. Sickle-cell anemia |
| Mutations in noncoding sequences | Mutation in components of an organism’s DNA that do not actually code protein sequences. | – PTF1A enhancer mutation can cause Pancreatic agenesis. – RET enhancer mutation can cause Hirshsprung disease. |
| Mutations in introns | Mutations in intervening noncoding segments of DNA. | A splice site mutation produces the parathyroid hormone. |
| Mutations in exons | Mutation in part of a gene that will encode a part of the final RNA. | Many issues. |
| Deletions | Deletion is a mutation that results in loss of genetic material. Deletions can be small or large. | -DiGeorge syndrome – 22q11 deletion. -Cri-du-chat syndrome – microdeletion on the short arm of chromosome 5. -Williams syndrome – microdeletion of long arm of chromosome 7. |
| Insertions | Addition of one or more nucleotide base pairs into a DNA sequence. | Huntington’s disease Fragile X syndrome |
| Trinucleotide repeats | Trinucleotide repeat expansions are a type of mutation where trinucleotide repeats in certain genes or introns exceed the normal, stable threshold. | – Fragile X syndrome – CGG repeats in fragile X mental retardation 1 (FMR1) gene. – Huntington’s disease – autosomal dominant trinucleotide CAG repeat expansion on chromosome 4. |
What are Mendelian Disorders?
Mendelian disorders are a single gene disorders, when a certain gene is known to cause a disease.
What are the Transmission Patterns of Single Gene Disorders?
The transmission patterns of single gene disorders are:
- Autosomal dominant
- Autosomal recessive
- X linked disorders
What are Autosomal Dominant Disorders?
Autosomal dominant disorders are disorders caused by mutations in dominant gene located on one of the nonsex chromosomes (autosomes).
What are Autosomal Recessive Disorders?
Autosomal recessive disorders are due to mutations in recessive gene located on one of the nonsex chromosomes.
What are X linked disorders?
X linked disorders are disorders caused by mutations in genes located on the X chromosome.
What are Single Gene Disorders?
Single gene disorders are caused by DNA changes in one particular gene, that often have predictable inheritance patterns. Examples of single gene disorders include cystic fibrosis, hemochromatosis, Tay-Sachs disease, and sickle cell anemia.
What are Disorders Associated with Defects in Structural Proteins?
Disorders associated with defects in structural proteins may be caused by incorrect protein folding.
What is Marfan Syndrome?
Marfan syndrome is an autosomal dominant connective tissue disorder due to defective fibrillin.
What is the Pathology of Marfan Syndrome?
The pathology of Marfan syndrome is:
-Etiology: The cause of Marfan syndrome is FBN1 gene mutation on chromosome 15.
-Genes involved: FBN1 gene mutation on chromosome 15.
-Pathogenesis: The sequence of events that lead to Marfan syndrome is defective fibrillin, a glycoprotein that forms a sheath around elastin.
How does Marfan Syndrome Present?
Marfan syndrome affects males and females equally, and the mutation shows no ethnic or geographical bias. The symptoms, features, and clinical findings associated with include tall stature, long extremities, pectus carinatum or pectus excavatum A, hypermobile joints, and long fingers.
How is Marfan Syndrome Diagnosed?
The diagnosis of Marfan syndrome relies on defined clinical criteria. The Ghent criteria, comprising a set of major and minor manifestations in different body systems. Chest x-ray, electrocardiogram (ECG) and echocardiogram are used to evaluate changes in the heart and blood vessels, and detect heart rhythm problems. CT or MRI is needed to evaluate for dural ectasia. Genetic testing is used to confirm diagnosis.
How is Marfan Syndrome Treated?
There is no cure for Marfan syndrome. Regular checkups are recommended to monitor the cardiovascular system to slow the progression of aortic dilation and prevent any damage to heart valves by eliminating heart arrythmias, by minimizing the heart rate, and lowering the person’s blood pressure.
What is the Prognosis of Marfan Syndrome?
The prognosis of Marfan syndrome is good. Adequate prophylactic monitoring and prophylactic therapy offers something approaching a normal lifespan, and more manifestations of the disease are being discovered as more patients live longer.
What is Ehlers-Danlos syndrome?
Ehlers-Danlos syndrome is a group of inherited connective tissue disorders caused by abnormalities in the structure, production, and/or processing of collagen.
What is the Pathology of Ehlers-Danlos Syndrome?
The pathology of Ehlers-Danlos syndrome is:
-Etiology: The cause of Ehlers-Danlos syndrome is mutations in a variety of genes, that may lead to abnormalities in the structure, production, and/or processing of collagen.
-Genes involved: Genes including COL5A1, COL5A2, COL1A1, COL3A1, TNXB, PLOD1, COL1A2, FKBP14 and ADAMTS2.
-Pathogenesis: The sequence of events that lead to Ehlers-Danlos syndrome are mutations in variety of genes leading to change the structure, production, and/or processing of collagen, or proteins that interact with collagen. A defect in collagen can weaken connective tissues in the skin, bones, blood vessels, and organs, resulting in the signs and symptoms of EDS.
How does Ehlers-Danlos syndrome Present?
Patients with Ehlers-Danlos syndrome typically affects either male or female, present at birth or in early childhood. The symptoms, features, and clinical findings associated with Ehlers-Danlos syndrome include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.
How is Ehlers-Danlos syndrome Diagnosed?
Ehlers-Danlos syndrome is diagnosed by medical history and clinical observation. The Beighton criteria are used to assess the degree of joint hypermobility. Diagnostic tests include collagen gene-variant testing, collagen typing via skin biopsy, echocardiogram, and lysyl hydroxylase or oxidase activity.
How is Ehlers-Danlos syndrome Treated?
Ehlers-Danlos syndrome is treated with supportive treatment.
What is the Prognosis of Ehlers-Danlos syndrome?
The prognosis of Ehlers-Danlos syndrome is dependent on the specific type of EDS they have.
What are Disorders Associated with Defects in Receptor Proteins?
Disorders associated with defects in receptor proteins are structural abnormalities of receptor proteins, that altered their function of receiving and transducing signals.
An example is familial hypercholesterolemia.
What is Familial Hypercholesterolemia?
Familial hypercholesterolemia is a genetic disorder characterized by high cholesterol levelswhich include very high levels of low-density lipoprotein in the blood and early cardiovascular disease.
What is the Pathology of Familial Hypercholesterolemia?
The pathology of familial hypercholesterolemia is:
-Etiology: The cause of familial hypercholesterolemia is mutation in the gene encoding the receptor for LDL.
-Genes involved: the most common genetic defects are LDLR mutations, ApoB mutations, PCSK9 mutations and LDLRAP1.
-Pathogenesis: The sequence of events that lead to Familial Hypercholesterolemia: LDL receptor function is reduced or absent.
How does Familial Hypercholesterolemia Present?
Patients with familial hypercholesterolemia typically either male or female present early in life. The symptoms, features, and clinical findings associated with familial hypercholesterolemia include yellow deposits of cholesterol-rich fat, xanthelasma palpebrarum, xanthoma, and cardiovascular disease.
How is Familial Hypercholesterolemia Diagnosed?
Familial Hypercholesterolemia is diagnosed by physical exams, personal or family history of hypercholesterolemia, early‐onset ASCVD, and the concentration of circulating LDL‐C.
How is Familial Hypercholesterolemia Treated?
Familial hypercholesterolemia is treated with statins, bile acid sequestrants, or other lipid-lowering agents. LDL apheresis may also be considered.
What is the Prognosis of Familial Hypercholesterolemia?
The prognosis of familial hypercholesterolemia is dependent on how closely an individual follows the lifestyle and diet recommendations.
What are Disorders Associated with Defects in Enzymes?
Disorders associated with defects in enzymes are inherited defects that result in a absence or abnormal functioning of enzymes.
What are Lysosomal Storage Diseases?
Lysosomal storage diseases are a group of over 70 rare inherited metabolic disorders that result from defects in lysosomal function.
Examples of lysosomal storage diseases include:
- Glycoogenosis
- Sulfatidoses
- Mucopolysaccharidoses
- Mucopolysaccharidoses
- Mucolipidoses
- Fucosidosis
- Mannosidosis
- Aspartglycosaminuria
- Wolman disease
- Acid phosphate deficiency
| Disease | Enzyme Deficiency | Major Accumulation Metabolites |
| Glycogenosis | Type 2 – Pompe disease Α-1,4-Glucosidase (lysosomal glucosidase) | Glycogen |
| Sphingolipidoses Gm1 gangliosidosis Type 1 – infantile, generalized Type 1 – juvenile Gm2 gangliosidosis Tay-Sachs disease Sandhoff disease Gm2 gangliosidosis variant AB | Gm1 ganglioside β-galactosidase Hexaminidase, α subunit Hexaminidase, β subunit Ganglioside activator protein | Gm1 ganglioside, galactose-containing oligosaccharides Gm2 ganglioside Gm2 ganglioside, globoside Gm2 ganglioside |
| Sulfatidoses Metachromatic leukodystrophy Multiple sulfatase deficiency Krabbe disease Fabry disease Gaucher disease Niemann-Pick disease | Arylsulfatase A Arylsulfatase A, B, C, steroid sulfatase; iduronate sulfatase; heparin N-sulfatase Galactocerebrosidase (galactosylceramidase) α-galactosidase A Glucocerebrosidase Sphingomyelinase | Cerebroside sulfate Sulfatide, steroid sulfate, heparin sulfate, dermatan sulfate Galactocerebroside, psychosine Ceramide trihexoside Glucocerebroside Sphingomyelin |
| Mucopolysaccharidoses Hurler syndrome Hunter syndrome | α-l-iduronidase Iduronate-2-sulfatase | Heparan sulfate, dermatan sulfate Heparan sulfate, dermatan sulfate |
| Mucolipidoses I-cell disease and pseudo-Hurler polydystrophy | Deficiency of phosphorylating enzymes essential for the formation of mannose-6-phosphate recognition marker; acid hydrolases lacking the recognition marker cannot be targeted to the lysosomes but are secreted extracellulary | Mucopolysaccharide, glycolipid |
| Other diseases of complex carbohydrates Fucosidosis Mannosidosis Aspartylglycosaminuria | α-Fucosidase α-Mannosidase Aspartylglycosamine amide hydrolase | Fucose-containing sphingolipids and glycoprotein fragments Mannose-containing oligosaccharides Aspartyl-2-deaxy-2-acetamido-glycosylamine |
| Other lysosomal storage disease Wolman disease Acid phosphate deficiency | Acid lipase Lysosomal acid phosphate | Cholesterol esters, triglycerides Phosphate esters |
What are Glycogen storage diseases?
Glycogen storage diseases are group of diseases resulting in abnormal glycogen metabolism and an accumulation of glycogen within cells.
Examples of glycogen storage disease include:
Types I,II,III,IV, and V.
| Types | Affected tissue | Enzyme defect | Clinical features | Tissue needed for diagnosis* | Outcome |
| Von Gierke disease (type I) | Liver, intestine, kidney | Glucose-6-phosphatase | Hepatomegaly, hypoglycemia, stunted growth, obesity, hypotonia | Liver | If patient survive initial hypoglycemia, prognosis is good; hyperuricemia is a late complication |
| Pompe disease (type II) | Liver, muscle, heart | Lysosomal acid α-1,4-glucosidase with α-1,6-glucosidase activity | Cardiomegaly, hypertrophic cardiomyopathy, hypotonia, exercise intolerance, and systemic findings. | Leukocytes, liver, muscle | Death in first 6 months; juvenile and adult variants seen |
| Forbes disease (type III) | Liver, muscle (abnormal glycogen structure) | Amylo-1, 6-dlucosidase | Hepatomegaly, hypoglycemia, stunted growth, obesity, hypotonia | Leukocytes, liver, muscle | Good prognosis |
| Anderson disease (type IV) | Liver, (abnormal glycogen structure) | 1,4-α-glucan branching enzyme | Failure to thrive, hepatomegaly, cirrhosis and its complications | Leukocytes, liver, muscle | Death in first 3 years |
| McArdle disease (type V) | Muscle only | Phosphorylase | Muscle cramps and myoglobinuria after exercise (in adults) | Muscle | Normal lifespan; exercise must be avoided |
What are Disorders Associated with Defects in Proteins That regulate Cell Growth?
Disorders Associated with Defects in Proteins That regulate Cell Growth are…
| Diseases | Inheritance | Features |
| Tay-Sachs | Recessive | Neurodegeneration, developmental delay, hyperreflexia, hyperacusis, “cherry-red” spot on macula A, lysosomes with onion skin. |
| Familial hypercholesterolemia | Dominant | Xanthelasma palpebrarum, tendon xanthoma, advanced atherosclerosis. |
| Sickle cell anemia | Recessive | Extravascular and intravascular hemolysis cause anemia. |
| Duchenne muscular dystrophy | X-linked Recessive | Weakness begins in pelvic girdle muscles and progresses superiorly. Pseudohypertrophy of calf muscles. |
| Cystic Fibrosis | Recessive | Recurrent pulmonary infections, pancreatic insufficiency, malabsorption with steatorrhea, fat-soluble vitamin deficiencies (A, D, E, K), biliary cirrhosis, liver disease, infertility in men and subfertility in women, nasal polyps, clubbing of nails. |
| Hemochromatosis | Recessive | Classic triad of cirrhosis, diabetes mellitus, skin pigmentation (“bronze diabetes”). |
| Huntington disease | Dominant | Chorea, athetosis, aggression, depression, dementia |
Complex Multigenic Disorders:
- Normal karyotype
- Structural abnormalities of chromosomes
- Chromosomal disorders
- Trisomies
- Trisome 21
- Trisomy 18
- Trisomy 13
- Trisomy 22
| Disease | Clinical Features |
| Down syndrome (trisomy 21) | Intellectual disability, prominent epicanthal folds, single palmar crease, flat facies, incurved 5th finger, gap between 1st 2 toes, duodenal atresia, Hirschsprung disease, congenital heart disease (atrial septal defect), and brushfield spots. Associated with early-onset Alzheimer disease (chromosome 21 codes for amyloid precursor protein), increase risk of AML/ALL. |
| Edwards syndrome (trisomy 18) | Prominent occiput, rocker-bottom feet, intellectual disability, clenched fists, overlapping fingers, low-set ears, micrognathia (small jaw), congenital heart disease, omphalocele, myelomeningocele. Death usually occurs by 1 year of age. |
| Patau syndrome (trisomy 13) | Intellectual disability, rocker-bottom feet, microphthalmia, microcephaly, cleft lip/palate, holoprosencephaly, polydactyly, cutis aplasia, congenital heart disease, polycystic kidney disease, omphalocele. Death usually occurs by 1 year of age. |
| Schmid–Fraccaro syndrome (trisomy 22) | Abnormalities of the eyes, ears, anal region, heart and/or kidney. |
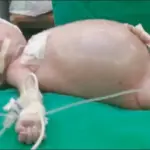
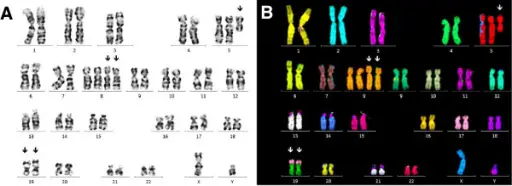
What is 22q11.2 Deletion syndrome?
22q11.2 deletion syndrome is a syndrome caused by a microdeletion on the long arm of chromosome 22 resulting in abnormal development of several body systems.
What is the Pathology of 22q11.2 Deletion syndrome?
The pathology of 22q11.2 deletion syndrome is:
-Etiology: The cause of 22q11.2 deletion syndrome is heterozygous deletion of part of the long arm (q) of chromosome 22, region 1, band 1, sub-band 2 (22q11.2).
-Genes involved: 1. Haploinsufficiency of the TBX1 gene (T-box transcription factor TBX1). Haploinsufficiency of the DGCR8 gene has been linked to improper regulation of the microRNA miR-338 and 22q11.2 deletion phenotypes. TANGO2 gene.
-Pathogenesis: 22q11.2 microdeletion causes to failure to develop 3rd and 4th pharyngeal pouches which leads to an absent thymus.
How does 22q11.2 Deletion syndrome Present?
Patients with 22q11.2 deletion syndrome typically affects either male or female present at birth. The symptoms, features, and clinical findings associated with 22q11.2 deletion syndrome include congenital heart disease, (ventricular septal defect, tetralogy of Fallot, interrupted aortic arch, and truncus arteriosus), palatal abnormalities, and immune deficiency.
How is 22q11.2 Deletion syndrome Diagnosed?
22q11.2 deletion syndrome diagnosis is established by identification of a heterozygous deletion at chromosome 22q11.2 on chromosomal microarray analysis or other genomic analyses.
How is 22q11.2 Deletion syndrome Treated?
22q11.2 deletion syndrome is treated in order to certain individual features using standard treatments. No cure is known for DiGeorge syndrome.
What is the Prognosis of 22q11.2 Deletion syndrome?
There is a wide range of symptoms and severity among people with 22q11.2 deletion syndrome. The long-term outlook for each person depends on the specific signs and symptoms each individual has.
What are Genetic disorders involving sex chromosomes?
Genetic disorders involving sex chromosomes include:
- Klinefelter syndrome
- Turner syndrome
- Hermaphroditism
- Pseudohermaphroditism
What is Klinefelter syndrome?
Klinefelter syndrome is a group of chromosomal disorders in which there is at least one extra X chromosome compared with the normal 46,XY male karyotype.
What is the Pathology of Klinefelter syndrome?
The pathology of Klinefelter syndrome is:
-Etiology: The cause of Klinefelter syndrome is retain of extra chromosome because of a nondisjunction event during paternal meiosis I, maternal meiosis I, or maternal meiosis II (gametogenesis). It is not an inherited condition.Maternal age is the only known risk factor. Women at 40 years have a four times higher risk for a child with Klinefelter syndrome than women aged 24 years.
-Genes involved: XXY.
-Pathogenesis: The sequence of events that lead to Klinefelter syndrome is congenital aneuploidy of the sex chromosomes.
How does Klinefelter syndrome Present?
Patients with Klinefelter syndrome typically males present at puberty. The symptoms, features, and clinical findings associated with Klinefelter syndrome include infertility and small, poorly functioning testicles. Often, symptoms are subtle and subjects do not realize they are affected. Sometimes, symptoms are more evident and may include weaker muscles, greater height, poor motor coordination, less body hair, breast growth, and less interest in sex. Often, these symptoms are noticed only at puberty. Intelligence is usually normal, but reading difficulties and problems with speech are more common.
How is Klinefelter syndrome Diagnosed?
Klinefelter syndrome is diagnosed by thorough physical examination. Testicular volume is measured by ultrasound examination, which usually also shows testicular hypoechogenicity. Smear testing can be used to look for Barr bodies in the buccal epithelium, corresponding to the inactive supernumerary X chromosome. Diagnosis is finally confirmed cytogenetically, by karyotyping.
How is Klinefelter syndrome Treated?
Klinefelter syndrome is treated with a number of treatments. Physical therapy, occupational therapy, speech and language therapy, counselling, and adjustments of teaching methods may be useful. Testosterone replacement may be used. About 50% of affected males have a chance of fathering children.
What is the Prognosis of Klinefelter syndrome?
The prognosis of Klinefelter syndrome is good. People with the condition have a nearly normal life expectancy.
What is Turner syndrome?
Turner syndrome is a genetic condition in which a female is partially or completely missing an X chromosome.
What is the Pathology of Turner syndrome?
The pathology of Turner syndrome is:
-Etiology: The cause of Turner syndrome is absence of one complete or partial copy of the X chromosome in some or all the cells. In most cases, Turner syndrome is a sporadic event, and for the parents of an individual with Turner syndrome the risk of recurrence is not increased for subsequent pregnancies. While most people have 46 chromosomes, people with TS usually have 45 in some or all cells.
-Genes involved: Monosomy X.
-Pathogenesis: The sequence of events that lead to Turner syndrome is caused by the absence of one set of genes from the short arm of one X chromosome. In patients with 45,X karyotype, about two thirds are missing the paternal X chromosome.
How does Turner syndrome Present?
Patients with Turner syndrome typically female present at any age range. Often, it is diagnosed at birth due to heart problems, an unusually wide neck or swelling of the hands and feet. The symptoms, features, and clinical findings associated with Turner syndrome include short stature, ovarian dysgenesis, shield chest, bicuspid aortic valve, coarctation of the aorta, lymphatic defects, horseshoe kidney, high-arched palate, shortened 4th metacarpals.
How is Turner syndrome Diagnosed?
Turner syndrome is diagnosed by amniocentesis or chorionic villus sampling during pregnancy (prenatal). Turner syndrome can be diagnosed postnatally at any age. A karyotyping is the test of choice to diagnose Turner syndrome.
How is Turner syndrome Treated?
Turner syndrome is treated with estrogen replacement therapy, and growth hormone. Most of these significant conditions are treatable with surgery. Reproductive technologies have also been used to help women with Turner syndrome become pregnant if they desire.
What is the Prognosis of Turner syndrome?
The prognosis of Turner syndrome is good. Patients with Turner syndrome have an increased mortality rate, three times greater than the general population. Cardiovascular disease due to coronary heart disease and stroke in older patients is a significant factor. Of the congenital cardiovascular disease, an aortic aneurysm is the largest cause. Patients also demonstrate an increase in mortality due to pneumonia, diabetes, epilepsy, liver disease, and kidney disease.
What is Hermaphroditism?
Hermaphroditism is an intersex condition in which an individual is born with both ovarian and testicular tissue.
What is the Pathology of Hermaphroditism?
The pathology of Hermaphroditism is:
-Etiology: The causes of Hermaphroditism are different types of gonads.
-Genes involved: SRY gene.
-Pathogenesis: The sequence of events that lead to Hermaphroditism coexistence of seminiferous tubules and ovarian follicles.
How does Hermaphroditism Present?
Patients with hermaphroditism typically present at birth. The symptoms, features, and clinical findings associated with (disease in lower case) include ambiguous genitalia.
How is Hermaphroditism Diagnosed?
Hermaphroditism is diagnosed by initial investigations include chromosome analysis and an ultrasound scan to check the internal reproductive organs. Patients who present later in life have higher differentiation of genitalia. Diagnosis requires careful anatomical assessment via imaging modalities and/or laparoscopy. Biochemical endocrine investigation, cytogenetic and molecular genetic tests are also useful.
How is Hermaphroditism Treated?
Hermaphroditism is treated with hormone replacement and psychological support.
What is the Prognosis of Hermaphroditism?
The prognosis of hermaphroditism is good. Patients usually have normal life expectancy.
What is Pseudohermaphroditism?
Pseudohermaphroditism is a condition in which an individual has a matching chromosomal and gonadal tissue (ovary or testis) sex, but mismatching external genitalia.
What is the Pathology of Pseudohermaphroditism?
The pathology of pseudohermaphroditism is:
-Etiology: The causes of pseudohermaphroditism are inability to synthesize estrogens from androgens, hormonal resistance due to androgen receptor dysfunction, or 5α-reductase deficiency.
-Genes involved: Y chromosome. SRY gene.
-Pathogenesis: Persistent müllerian ducts.
How does Pseudohermaphroditism Present?
Patients with pseudohermaphroditism typically present early in life. In some cases, external sex organs associated with pseudohermaphroditism appear intermediate between a typical clitoris and penis. Thus, pseudohermaphroditism is sometimes not identified until puberty or adulthood. The symptoms, features, and clinical findings associated with pseudohermaphroditism include external genitalia of the opposite sex.
How is Pseudohermaphroditism Diagnosed?
Pseudohermaphroditism is diagnosed by initial investigations include chromosome analysis and an ultrasound scan to check the internal reproductive organs. Patients who present later in life have higher differentiation of genitalia. Diagnosis requires careful anatomical assessment via imaging modalities and/or laparoscopy. Biochemical endocrine investigation, cytogenetic and molecular genetic tests are required.
How is Pseudohermaphroditism Treated?
Hormonal and other medical interventions performed to modify atypical or ambiguous genitalia and other sex characteristics.
What is the Prognosis of Pseudohermaphroditism?
The prognosis of pseudohermaphroditism is good. Patients usually have normal life expectancy.
| Disease | Genetic Issue | Clinical Features |
| Klinefelter syndrome | X extra chromosome | Infertility and small, poorly functioning testicles. |
| Pure gonadal dysgenesis | None. | Impaired development of the gonads. |
| XYY male | Extra Y chromosome (47XYY) | Taller than average height, low muscle tone, or muscle weakness, clinodactyly, widely spaced eyes, behavioral disorders. |
| XXX female | Extra X chromosome (47XXX) | Learning disabilities, mild dysmorphic features such as hypertelorism and clinodactyly, early menopause, and increased height. |
| Mixed gonadal dysgenesis | 45,XO/46,XY mosaicism | Asymmetry in gonadal development of testis and streak gonad. |
| Turner syndrome | Completely missing second X chromosome (45,X or 45,X0) | Short stature, streak ovary, shield chest, bicuspid aortic valve, coarctation of the aorta, lymphatic defects (result in webbed neck or cystic hygroma; lymphedema in feet, hands), horseshoe kidney, high-arched palate, shortened 4th metacarpals. |
What are Trinucleotide-Repeat Disorders?
Trinucleotide-repeat disorders are disorders caused by trinucleotide repeat expansion.
Examples of trinucleotide-repeat disorders include:
- Fragile X syndrome
- Fragile X tremor ataxia
- Hunington’s disease
- Myotonic dystrophy
- Spinobulbar muscular atrophy
- Spinocerebellar ataxia
- Dentatorubral-pallidoluysian atrophy
What is Fragile X syndrome?
Fragile X syndrome is a genetic disorder characterized by mild-to-moderate intellectual disability.
What is the Pathology of Fragile X syndrome?
The pathology of Fragile X syndrome is:
-Etiology: The cause of Fragile X syndrome is a mutation of the fragile X mental retardation 1 (FMR1) gene on the X chromosome, most commonly an increase in the number of CGG trinucleotide repeats in the 5′ untranslated region of FMR1.
-Genes involved: Fragile X mental retardation gene on X chromosome.
-Pathogenesis: The sequence of events that lead to Fragile X syndrome: expansion of the CGG triplet repeat within the FMR1 (fragile X mental retardation 1) gene on the X chromosome. This results in silencing (methylation) of this part of the gene and a deficiency of the resultant protein (FMRP), which is required for the normal development of connections between neurons. FMRP is found throughout the body, but in highest concentrations within the brain and testes. The downregulation of GABA pathways, which serve an inhibitory function and are involved in learning and memory, may be a factor in the anxiety symptoms which are commonly seen in FXS.
How does Fragile X syndrome Present?
Patients with Fragile X syndrome typically either male or female, but females with full FMR1 mutations may have a milder phenotype than males due to variability in X-inactivation, present at childhood. The symptoms, features, and clinical findings associated with Fragile X syndrome include intellectual disability, post-pubertal macroorchidism(enlarged testes), long face with a large jaw, large everted ears, autism, and mitral valve prolapse. .
How is Fragile X syndrome Diagnosed?
Fragile X syndrome is diagnosed using DNA testing for fragile X syndrome. Karyotyping may reveal other chromosomal anomalies, and both a standard karyotype and DNA testing are suggested when a possible diagnosis of fragile X syndrome is considered.
How is Fragile X syndrome Treated?
Fragile X syndrome is treated with speech therapy, behavioral therapy, occupational therapy, special education, or individualised educational plans, and, when necessary, treatment of physical abnormalities. Persons with fragile X syndrome in their family histories are advised to seek genetic counseling to assess the likelihood of having children who are affected, and how severe any impairments may be in affected descendants.
What is the Prognosis of Fragile X syndrome?
The prognosis of Fragile X syndrome is fair. Life expectancy for FXS is 12 years lower than the general population and the causes of death are similar to those found for the general population.
What is Fragile X tremor ataxia?
Fragile X tremor ataxia is a late-onset neurodegenerative disorder most frequently seen in male premutation carriers of Fragile X syndrome (FXS) over 50 years old.
What is the Pathology of Fragile X tremor ataxia?
The pathology of Fragile X tremor ataxia is:
-Etiology: The cause of Fragile X tremor ataxia is a trinucleotide repeat expansion of 50-200 CGG repeats in the Fragile X mental retardation-1 (FMR1) gene.
-Genes involved: Fragile X mental retardation-1 (FMR1) gene.
-Pathogenesis: The pathogenesis of FXTAS results from the direct neural cell toxicity of elevated levels of the expanded-CGG-repeat FMR1 mRNA (RNA toxic gain-of-function), which leads to dysregulation of a number of proteins including lamin A/C and alpha B crystallin.
How does Fragile X tremor ataxia Present?
Patients with Fragile X tremor ataxia typically males present at age range of over 50. The symptoms, features, and clinical findings associated with Fragile X tremor ataxia include an intention tremor, cerebellar ataxia, and parkinsonism.
How is Fragile X tremor ataxia Diagnosed?
Fragile X tremor ataxia is diagnosed using a combination of molecular, clinical, and radiological findings. Definite diagnosis is made using a major radiological finding and one major clinical finding must be present.
How is Fragile X tremor ataxia Treated?
Fragile X tremor ataxia is treated with medications for alleviating symptoms of tremor, ataxia, mood changes, anxiety, cognitive decline, dementia, neuropathic pain, or fibromyalgia. Neurological rehabilitation has not been studied for patients with FXTAS but should also be considered as a possible form of therapy. Additionally, occupational and physical therapy may help to improve function. There is no treatment modality aimed at reversing the pathology of FXTAS.
What is the Prognosis of Fragile X tremor ataxia?
The prognosis of Fragile X tremor ataxia varies widely between each case. The onset of symptoms may be gradual, with progression of the disease spanning multiple years or decades. Alternatively, symptoms may progress rapidly.
What is Hunington’s disease?
Hunington’s disease is neurodegenerative disorder of the central nervous system characterized by unwanted choreatic movements, behavioral and psychiatric disturbances and dementia.
What is the Pathology of Huntington’s disease?
The pathology of Hunington’s disease is:
-Etiology: The cause of Hunington’s disease is autosomal dominant trinucleotide (CAG)repeat expansion in the huntingtin (HTT) gene on chromosome 4.
-Genes involved: huntingtin (HTT) gene on chromosome 4.
-Pathogenesis: The sequence of events that lead to Huntington’s disease is expansion of CAG repeats of cytosine-adenine-guanine in the gene coding for the Huntington protein. Damage may occur in the subcortical basal ganglia, initially in the striatum, but as the disease progresses, other areas of the brain are also affected, including regions of the cerebral cortex. Early symptoms include abnormal movements, and mood changes.
How does Hunington’s disease Present?
Patients with Hunington’s disease typically are males over forty years old. The symptoms, features, and clinical findings associated with Huntington’s disease include chorea, athetosis, aggression, depression, dementia.
How is Hunington’s disease Diagnosed?
Hunington’s disease is diagnosed basing on physical exam. Genetic testing can be used to confirm a physical diagnosis if no family history of HD exists. Even before the onset of symptoms, genetic testing can confirm if an individual or embryo carries an expanded copy of the trinucleotide repeat (CAG) in the HTT gene that causes the disease. Genetic counseling is available to provide advice and guidance throughout the testing procedure and on the implications of a confirmed diagnosis.
How is Hunington’s disease Treated?
Hunington’s disease treatment inoves management of symptoms which include dopamine blockers. Anxiolytic and antidepressant therapy may be necessary.
What is the Prognosis of Hunington’s disease?
The prognosis of Hunington’s disease is fair. Life expectancy is around 20 years following the onset of visible symptoms.
What is Myotonic dystrophy?
Myotonic dystrophy is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness.
What is the Pathology of Myotonic Dystrophy?
The pathology of myotonic dystrophy is:
-Etiology: The microsatellite expansion responsible for DM1 is of cytosine-thymine-guanine (CTG) triplet repeats.
-Genes involved: DMPK gene in myotonic dystrophy type 1 on the long arm of chromosome 19, CNBP gene on chromosome 3 in myotonic dystrophy type 1.
-Pathogenesis: The sequence of events that lead to Myotonic dystrophy involves unusually long messenger RNA that forms clumps inside the cell that interfere with the production of many other proteins. These changes prevent muscle cells and cells in other tissues from functioning normally.
How does Myotonic dystrophy Present?
Patients with myotonic dystrophy typically affects either male or female present at age range of 20s to 30s. The symptoms, features, and clinical findings associated with myotonic dystrophy include muscle weakness, early onset of cataracts, and myotonia, which is delayed relaxation of muscles after contraction. Other organs affected include the heart, lungs, gastrointestinal tract, skin, and brain. Insulin resistance may also occur.
How is Myotonic Dystrophy Diagnosed?
Myotonic dystrophy is diagnosed by doing a physical exam. Laboratory tests for muscle enzymes can be used to clarify the clinical diagnosis of myotonic dystrophy. Electromyography may also be beneficial. Muscle biopsy and molecular testing are confirmatory.
How is Myotonic Dystrophy Treated?
Myotonic dystrophy is treated with mexiletine or carbamazepine which can help relax muscles. NSAIDs, braces, and wheelchairs may be needed as the disease progresses.
What is the Prognosis of Myotonic Dystrophy?
The prognosis of myotonic dystrophy is poor. Life expectancy in non-congenital late-onset or adult onset DM1 is in the early 50s, with pulmonary complications being the leading cause of death, followed by cardiac complications.
What is Spinobulbar Muscular Atrophy?
Spinobulbar muscular atrophy is a progressive debilitating neurodegenerative disorder that causes muscle cramps and progressive weakness.
What is the Pathology of Spinobulbar Muscular Atrophy?
The pathology of spinobulbar muscular atrophy is:
-Etiology: The cause of spinobulbar muscular atrophy is expansion of a CAG repeat in the first exon of the androgen receptor gene.
-Genes involved: AR gene, located in the X chromosome. CAG trinucleotide repeats.
-Pathogenesis: The sequence of events that lead to spinobulbar muscular atrophy involves expansion of a CAG repeat in the first exon of the androgen receptor gene. The repeat expansion likely causes a toxic gain of function in the androgen receptor protein, which leads to degeneration of motor neurons in the brain stem and spinal cord.
How does Spinobulbar Muscular Atrophy Present?
Patients with spinobulbar muscular atrophy typically affects males and is very rare in females, present at age range of 30 to 50 years old. The symptoms, features, and clinical findings associated with spinobulbar muscular atrophy include muscle weakness, muscle atrophy, and fasciculations. Affected individuals may have gynecomastia, testicular atrophy, and reduced fertility.
How is Spinobulbar Muscular Atrophy Diagnosed?
Spinobulbar muscular atrophy is diagnosed by identifying the number of CAG repeats in the AR gene using molecular techniques such as PCR.
How is Spinobulbar Muscular Atrophy Treated?
Spinobulbar muscular atrophy is treated by supportive treatment. Surgery may achieve correction of the spine, and early surgical intervention should be done in cases where prolonged survival is expected.
What is the Prognosis of Spinobulbar Muscular Atrophy?
The prognosis of spinobulbar muscular atrophy is fair. There is a shortened life expectancy.
What is Spinocerebellar Ataxia?
Spinocerebellar ataxia is a genetically heterogeneous group of inherited progressive degenerative disorders, the clinical hallmark of which is loss of balance and coordination accompanied by slurred speech.
What is the Pathology of Spinocerebellar Ataxia?
The pathology of spinocerebellar ataxia is:
-Etiology: The cause of spinocerebellar ataxia is mutations in many different genes cause the different types of spinocerebellar ataxia (SCA).
-Genes involved: ATXN1 gene mutations that cause SCA1 involve a DNA segment known as a CAG trinucleotide repeat.
-Pathogenesis: The sequence of events that lead to spinocerebellar ataxia are reduction in mass of frontal, temporal, and parietal portions of the brain along with the cerebellar peduncles, brainstem, and cranial nerve III.
How does Spinocerebellar Ataxia Present?
Patients with spinocerebellar ataxia are typically males. The symptoms, features, and clinical findings associated with Spinocerebellar ataxia include incoordination of gait and poor coordination of hands, speech, and eye movements, parkinsonism, chorea, pyramidalism, cognitive impairment, peripheral neuropathy, and seizures.
How is Spinocerebellar Ataxia Diagnosed?
Spinocerebellar ataxia is diagnosed with genetic testing.
How is Spinocerebellar Ataxia Treated?
Spinocerebellar ataxia has no cure. Zolpidem or varenicline may improve symptoms.
What is the Prognosis of Spinocerebellar Ataxia?
The prognosis of spinocerebellar ataxia is fair.
What is Dentatorubral-Pallidoluysian Atrophy?
Dentatorubral-pallidoluysian atrophy is a rare autosomal dominant neurodegenerative disorder clinically characterized by various combinations of cerebellar ataxia, choreoathetosis, myoclonus, epilepsy, dementia, and psychiatric symptoms.
What is the Pathology of Dentatorubral-Pallidoluysian Atrophy?
The pathology of Dentatorubral-pallidoluysian atrophy is:
-Etiology: The cause of dentatorubral-pallidoluysian atrophy is unstable expansion of CAG repeats coding for polyglutamine stretches located in exon 5 of the DRPLA gene.
-Genes involved: Atrophin-1 gene.
-Pathogenesis: The sequence of events that lead to dentatorubral-pallidoluysian atrophy are due to unstable expansion of CAG repeats coding for polyglutamine stretches located in exon 5 of the DRPLA gene. Dentatorubral-pallidoluysian is characterized by marked, generalized brain atrophy and the accumulation of atrophin-1 with expanded glutamine stretches. Mutant atrophin-1 proteins have been found in neuronal intranuclear inclusions (NII) and diffusely accumulated in the neuronal nuclei. While the role of NIIs (pathologic or protective) is unclear, the diffuse accumulation of mutant protein is regarded as toxic.
How does Dentatorubral-Pallidoluysian Atrophy Present?
Patients with dentatorubral-pallidoluysian typically affects either male or female present at varying age ranges. The symptoms, features, and clinical findings associated with dentatorubral-pallidoluysian atrophy include myoclonus, seizures of different types, behavioral changes, progressive intellectual disability and deterioration, and ataxia. When it begins after the age of 20 years it presents with: ataxia, choreoathetosis or dystonia, delusions, and dementia.
How is Dentatorubral-Pallidoluysian Atrophy Diagnosed?
Dentatorubral-pallidoluysian atrophy is diagnosed based on positive family history, clinical findings, and genetic testing. To quantify the extent of the disease, an MRI, EEG and neuropsychological testing are recommended.
How is Dentatorubral-Pallidoluysian Atrophy Treated?
Dentatorubral-pallidoluysian atrophy is treated with anticonvulsants if seizures are present. Psychotropic medications may help with psychiatric disturbances. Physical therapy has also been recommended to maintain function as the condition progresses and occupational therapy to focus on activities of daily living, advice for carers and adaptation to the environment.
What is the Prognosis of Dentatorubral-Pallidoluysian Atrophy?
The prognosis of Dentatorubral-pallidoluysian atrophy is poor. Seizures and dysphagia with frequent fluid and food aspiration cause bronchopneumonia and subsequent death.
| TRD | Mode of inheritance | Repeats involved | Examples of presentation |
| Dentatorubral-pallidoluysian atrophy | Autosomal dominant | CAG≥49 Chromosome 12 | Symptom onset is variable and depends on trinucleotide repeat length: can present with ataxia, dementia, depression, anxiety, chorea, etc. |
| Fragile X syndrome | X-linked dominant | CGG>200 | Intellectual disability, epilepsy, attention-deficit hyperactivity disorder, autistic-like features. |
| Fragile X tremor ataxia | X-linked dominant | CGG >50 | Problems of movement with cerebellar gait ataxia and action tremor. |
| Friedreich`s ataxia | Autosomal recessive | GAA≥70 Chromosome 9 | Progressive ataxia, dysarthria, depression, etc. |
| Hunington’s disease | Autosomal dominant | CAG≥40 Chromosome 4 | Chorea, dementia, depression. |
| Myotonic dystrophy | Autosomal dominant | CTG>50 Chromosome 19 | Intellectual disability or psychiatric symptoms can occur but main presentation is usually with muscle weakness, etc. |
| Spinobulbar muscular atrophy | X-linked recessive | CAG≥38 | Progressive muscles weakness and atrophy, fasciculation, etc. |
| Spinocerebellar ataxia | Autosomal recessive | Various, depending on the subtype | Variability in age of onset dependent on trinucleotide repeat length. Psychosis and dementia can occur with some subtypes, but progressive ataxia is the main presentation. |
What are Mutations in Mitochondrial Genes?
Mutations in mitochondrial genes are mutations that occur in the circular mitochondrial genes.
Mutations in Mitochondrial Genes include:
- MELAS
- Leber hereditary optic neuropathy
What is MELAS?
Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) is a multisystem and progressive neurodegenerative disorder.
What is the Pathology of MELAS?
The pathology of MELAS is:
-Etiology: The cause of MELAS is by mutations in mitochondrial DNA.
-Genes involved: can result from mutations in one of several genes, including MT-ND1, MT-ND5, MT-TH, MT-TL1, and MT-TV.
-Pathogenesis: The sequence of events that lead to MELAS impaired mitochondrial translation and protein synthesis including the mitochondrial electron transport chain which impair energy production.
How does MELAS Present?
Patients with MELAS typically affects either male or female, present at age range of 3-15 years old. The symptoms, features, and clinical findings associated with MELAS include hypertrophic cardiomyopathy, seizures, stroke-like episodes, diabetes, hearing loss, ptosis, and epilepsy. Patients may also experiance muscle fatigue and pain, generalized myopathy, myalgia and severe headache.
How is MELAS Diagnosed?
MELAS is diagnosed based on clinical findings and molecular genetic testing. Lactate and pyruvate concentrations and CSF protein may be helpful. Brain MRI may be used to look for stroke-like lesions. Electrocardiogram may be used to check for heart rhythm abnormalities and echocardiogram may be used to diagnose cardiomyopathy. Muscle biopsy may show ragged red fibers.
How is MELAS Treated?
MELAS is treated with enzymes, amino acids, antioxidants and vitamins
What is the Prognosis of MELAS?
The prognosis of MELAS is poor.
What is Leber hereditary optic neuropathy?
Leber hereditary optic neuropathy is s an inherited form of bilateral optic atrophy in which the primary etiological event is a mutation in the mitochondrial genome.
What is the Pathology of Leber hereditary Optic Neuropathy?
The pathology of Leber hereditary optic neuropathy is:
-Etiology: The cause of Leber hereditary optic neuropathy is a mutation at mtDNA nucleotide.
-Genes involved: mutations in the MT-ND1, MT-ND4, MT-ND4L, and MT-ND6 genes cause Leber hereditary optic neuropathy.
-Pathogenesis: The sequence of events that lead to Leber hereditary optic neuropathy are mutations in mitochondrial DNA that can alter the function of the mitochondria.
How does Leber Hereditary Optic Neuropathy Present?
Patients with Leber hereditary optic neuropathy are typically males about five times more than females. Patients tend to present at age range of 15-55 years old. The symptoms, features, and clinical findings associated with Leber hereditary optic neuropathy include decreased visual acuity, loss of color vision and a central scotoma.
How is Leber Hereditary Optic Neuropathy Diagnosed?
Leber hereditary optic neuropathy is diagnosed based on ophthalmologic findings which involves dilated fundus examination to identify characteristic changes in the optic disc and vascular changes during the acute phase, visual fields, electrophysiologic studies, and imaging, particularly OCT. Molecular genetic testing for mitochondrial genes associated with LHON can be used to confirm diagnosis.
How is Leber Hereditary Optic Neuropathy Treated?
Leber hereditary optic neuropathy is treated with supportive management and treatment through the usage of visual aids, occupational rehabilitation, and local social services.
What is the Prognosis of Leber Hereditary Optic Neuropathy?
The prognosis of Leber hereditary optic neuropathy is poor.
What is Genomic Imprinting?
Genomic imprinting is an epigenetic phenomenon that causes genes to be expressed in a parent-of-origin-specific manner. One gene copy of the gene is silenced by methylation, and the other copy of the gene is expressed.
What is Uniparental Disomy?
Uniparental disomy occur when both homologous chromosomes are inherited from one parent.
What is Defective Imprinting?
Defective imprinting is deletion of the active allele with inactivated other allele by methylation.
Examples of genomic imprinting include:
- Prader-Willi syndrome
- Angelman syndrome
What is Prader-Willi Syndrome
Prader-Willi Syndrome is is a genetic disorder caused by a loss of function of specific genes on chromosome 15.
What is the Pathology of Prader-Willi Syndrome?
The pathology of Prader-Willi Syndrome is:
-Etiology: The cause of Prader-Willi Syndrome is loss of imprinted genomic material within the paternal 15q11.2-13 locus.
-Genes involved: SNRPN gene, P gene (type II oculocutaneous albinism), UBE3A gene, and necdin gene
-Pathogenesis: The sequence of events that lead to Prader-Willi Syndrome are loss of expression of several genes encoded on the proximal long arm of chromosome 15.
How does Prader-Willi Syndrome Present?
Patients with Prader-Willi Syndrome typically are male or female, and typically present at early age. The symptoms, features, and clinical findings associated with Prader-Willi Syndrome include overeating, obesity, decreased muscular tone in infancy, mental retardation, small hands and feet, diabetes, and obesity.
How is Prader-Willi Syndrome Diagnosed?
Prader-Willi syndrome is diagnosed based on specific clinical features, and it is confirmed by genetic testing.
How is Prader-Willi Syndrome Treated?
Prader-Willi syndrome is treated with growth hormone to improve lean body mass, corrects osteopenia, does not appear to enhance the development of scoliosis, and anecdotally modulates behavior in some patients. Sex hormones may improve secondary sex characteristics.
What is the Prognosis of Prader-Willi Syndrome?
The prognosis of Prader-Willi Syndrome is good with proper treatment and lifestyle management.
What is Angelman Syndrome?
Angelman syndrome is a complex genetic disorder that primarily affects the nervous system.
What is the Pathology of Angelman Syndrome?
The pathology of Angelman syndrome is:
-Etiology: The cause of Angelman syndrome is deficiency of the E3 ubiquitin protein ligase (UBE3A) gene expression.
-Genes involved: UBE3A gene on maternal copy of chromosome 15.
-Pathogenesis: The sequence of events that lead to Angelman syndrome is a lack of expression of a gene UBE3A during development. Paternally derived UBE3A is silenced. Disease occurs when the maternal allele is deleted or mutated.
How does Angelman Syndrome Present?
Patients with Angelman syndrome may be male or female, and are typically younger. The symptoms, features, and clinical findings associated with Angelman syndrome include seizures, ataxia, severe intellectual disability, inappropriate laughter.
How is Angelman Syndrome Diagnosed?
Angelman syndrome is diagnosed based on a history of delayed motor milestones and then later a delay in general development, unusual movements, frequent laughing, hand flapping, and a wide-based gait. Genetic testing for deletion or inactivity on chromosome 15 by array comparative genomic hybridization (aCGH) or by BACs-on-Beads technology may be helpful.
How is Angelman Syndrome Treated?
Angelman syndrome is treated with supportive care.
What is the Prognosis of Angelman Syndrome?
The prognosis of Angelman syndrome is fair. People with Angelman syndrome appear to have a reduced but near-normal life expectancy, dying on average 10 to 15 years earlier than the general population.
| Prader-Willi syndrome | 15q11-15q13 (deletion of paternal allele | Overeating, obesity, decreased muscular tone in infancy, mental retardation, small hands and feet; obesity-related complications can decrease lifespan |
| Angelman syndrome | 15q11-15q13 (deletion of maternal allele) | Happy puppet, jerky movement, happy mood, unprovoked laughter, mental retardation, seizures |
What is Gonadal Mosaicism?
Gonadal mosaicism is a type of genetic mosaicism where more than one set of genetic information is found specifically within the gamete cells.
What is the Pathology of Gonadal Mosaicism?
The pathology of gonadal mosaicism is:
-Etiology: The cause of gonadal mosaicism is either a mutation that occurs after conception, or epigenetic regulation that alters DNA methylation.
-Genes involved: None.
-Pathogenesis: The sequence of events that lead to gonadal mosaicism may be autosomal or germline mosaicism.
How does Gonadal Mosaicism Present?
Depends on the disease.
How is Gonadal Mosaicism Diagnosed?
Gonadal mosaicism is diagnosed by minimally invasive procedures, such as blood sampling or amniotic fluid sampling. Genetic testing on collected samples may be helpful.
How is Gonadal Mosaicism Treated?
Depends on the disease.
What is the Prognosis of Gonadal Mosaicism?
Depends on the disease.
What is Molecular Genetic Diagnosis?
Molecular genetic diagnosis is a collection of techniques used to analyze biological markers in the genome and proteome, and how their cells express their genes as proteins, applying molecular biology to medical testing.
What are Diagnostic Methods and Indications for Genetic Testing?
Diagnostic Methods:
- Polymerase chain reaction (PCR)
- DNA Sequencing (Sanger sequencing, next-generation sequencing)
- Cytogenetics (Karyotyping and FISH); microarray testing
- Gene expression profiling
Indications for genetic tests are typically for conformational diagnosis, or to determine genetic risk.
What is Polymerase Chain Reaction Testing?
Polymerase chain reaction testing is a method widely used to rapidly make millions to billions of copies of a specific DNA sample to see what genes are present.
What is Molecular Analysis of Genomic Alterations?
Molecular analysis of genomic alterations is used to profile gene expression levels of thousands of genes at the same time to study certain diseases and treatments. This is able to detect single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) for a variety of applications including genotyping, clinical genetic testing, forensic analysis, cancer mutations, and genetic linkage analysis.
Examples of molecular analysis of genomic alterations include:
- Fluorescence in situ hybridization (FISH)
- Multiplex ligation-dependent probe amplification (MLPA)
- Southern blotting
- Cytogenomic array technology
What is Fluorescence in Situ Hybridization (FISH)?
Fluorescence in situ hybridization (FISH) is a laboratory technique for detecting and locating a specific DNA sequence on a chromosome. FISH relies on exposing chromosomes to a small DNA sequence (a probe) with an attached fluorescent molecule attached to it. The probe sequence binds to its corresponding sequence on the chromosome.
What is Multiplex Ligation-Dependent Probe Amplification (MLPA)?
Multiplex ligation-dependent probe amplification (MLPA) is a variation of the multiplex polymerase chain reaction that permits amplification of multiple targets with only a single primer pair. Multiplex ligation dependent probe amplification detects copy number changes at the molecular level. Specialized software programs are used for analysis. Identification of duplications or deletions may indicate pathogenic mutations.
What is Southern Blotting?
Southern blotting is a laboratory technique used to detect specific DNA molecules. Southern blots can be used to analyze an organism’s total DNA, also known as its genome, in order to identify a specific sequence of interest.
What is Cytogenomic Array Technology?
Cytogenomic array technology is microarray-based technologies for the investigation of specific loci and the entire genome.
What are Polymorphic Markers?
Polymorphic markers are a gene or DNA sequence with a known location on a chromosome that can be used to identify individuals or species.
What is Molecular Diagnosis?
Molecular diagnosis is a collection of techniques that are used to analyze genetics of biological specimens.
What is Genome-Wide analysis?
Genome-wide analysis is a genetic test that can scan the entire genome to find specific genetic patterns.
What are Epigenetic Alterations?
Epigenetic alterations are environmentally induced changes in the chemical structure or expression of DNA that does not change the DNA coding sequence. Epigenetic alterations may occur when methyl groups are added to or removed from DNA, or when changes are made to histones. These changes may occur with age and exposure to environmental factors, such as exercise, change in diet, and exposure to sunlight.
What is RNA Analysis?
RNA analysis is a multistage process that includes cloning, physical mapping, subcloning, sequencing, and information analysis of an RNA sequence.
What is Bioinformatics?
Bioinformatics is a subdiscipline of biology and computer science focused on analysis, acquisition, storage, and dissemination of biological information, especially DNA and proteomics.
What is Next Generation Sequencing?
Next generation sequencing is technology that allows rapid study of genomics and molecular biology.