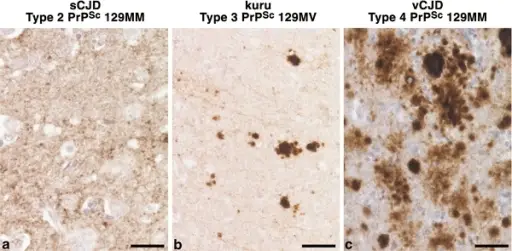
Distinct patterns of PrP deposition in brain in human prion disease. The disease aetiology, PRNP codon 129 genotype of the patient (M methionine, V valine) and the type of PrPSc detected in each sample (using the London classification of human PrPSc types [54]) is designated above each brain sample. The most common subtype of sporadic CJD (a) typically shows a diffuse, synaptic pattern of abnormal PrP deposition, while kuru (b) shows not only variably diffuse PrP deposition but also striking formation of PrP plaques in various areas of the brain. These PrP plaques are consistently distinct from those seen in vCJD (c), where PrP plaques are often surrounded by conspicuous vacuolation, designated ‘florid plaques’. Scale barsa, c 25 μm; b 50 μm.Molecular pathology of human prion disease.
Wadsworth JD, Collinge J - Acta neuropathologica (2010). Not Altered. CC.
Prion diseases are or transmissible spongiform encephalopathies that can cause rare progressive neurodegenerative disorders that affect both humans and animals. Prion diseases are usually rapidly progressive and fatal.
Examples of prion diseases include:
- Creutzfeldt-Jakob diseases
- Kuru
- Gerstman-Straussler-Scheinker syndrome
- Fatal familial insomnia