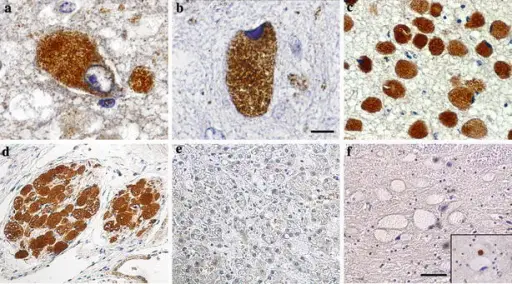
Immunodetection of ubiquitin in prosaposin deficiency (except for panel f). a Strong granular staining in perikarya of subcortical storage neurons in case 1 (day 27), and b case 2 (day 89). c Uniform strong ubiquitination in pontine storage neurons in case 3 (day 119), and d in peripheral neurons of the kidney hilus in the same case. e Absence of detectable ubiquitination in the storage cells in the liver in pSap deficiency (case 2). f Absence of ubiquitination in storage neurons in Niemann–Pick-disease type A. Inset shows intense ubiquitination of the small dystrophic axon close to another negative neuronal perikaryon. Scale bar 25 μm (a,b) and 100 μm (c–f). Neurolysosomal pathology in human prosaposin deficiency suggests essential neurotrophic function of prosaposin.
Sikora J, Harzer K, Elleder M - Acta neuropathologica (2006). Not Altered. CC.
Neuronal storage diseases are storage diseases in the central nervous system that result from a deficiency of a specific degradative lysosomal enzyme causing the accumulation of a substrate that is stored in the cytoplasm of the neuronal cell body, and occasionally in glia, macrophages, and the cells of other organs.
Examples of neuronal storage diseases include:
- Niemann-Pick disease
- Tay-Sachs disease