Cystic fibrosis is an inherited disorder that causes severe damage to the lungs, digestive system and other organs in the body. Cystic fibrosis affects the cells that produce mucus, sweat and digestive fluids.
What is the Pathology of Cystic Fibrosis?
The pathology of cystic fibrosis is:
-Etiology: The cause of cystic fibrosis is mutations in a gene called the cystic fibrosis transmembrane conductance regulator CFTR gene.
-Genes involved: CFTR gene mutation.
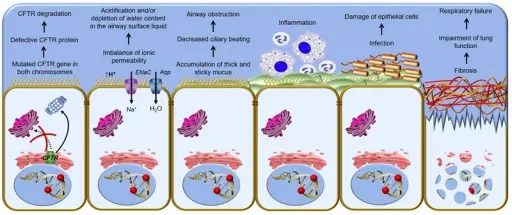
-Pathogenesis: The sequence of events that lead to cystic fibrosis are: mutation in gene causes production of a protein that does not fold normally and is not appropriately transported to the cell membrane, resulting in its degradation.
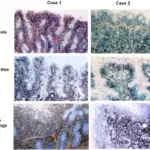
-Histology: The histology associated with cystic fibrosis shows hyperplasia of mucous cells, increased numbers of pulmonary neuroendocrine and indeterminate cells, degeneration and sloughing of epithelial cells.
How does Cystic Fibrosis Present?
Patients with cystic fibrosis typically present during childhood. The diagnosis after 30 to 60 years is rare. The symptoms, features, and clinical findings associated with cystic fibrosis include salty-tasting skin, persistent coughing, frequent lung infections, poor weight gain in spite of a good appetite, and frequent greasy bulky stools. Nasal polyps may also be present.
How is Cystic Fibrosis Diagnosed?
Cystic fibrosis is diagnosed by sweat test, genetic tests, blood sample to check for higher levels of a chemical called immunoreactive trypsinogen.
How is Cystic Fibrosis Treated?
Cystic fibrosis is treated by antibiotics to treat and prevent lung infections. Anti-inflammatory medications to lessen swelling in the airways in your lungs. Mucus-thinning drugs, such as hypertonic saline, to help you cough up the mucus, which can improve lung function.
What is the Prognosis of Cystic Fibrosis?
The prognosis of cystic fibrosis is not good. It tends to get worse over time and can be fatal if it leads to a serious infection or the lungs stop working properly.