Microcytic Anemias
Microcytic Anemia Pathology Video
Anemia with MCV < 80 fL/m3
Microcytic anemias are due to decreased production of hemoglobin.
Red blood cell (RBC) progenitor cells (erythroblasts) in the bone marrow are large and normally divide multiple times to produce smaller mature cells (MCV = 80 -100 micrometers3).
An additional division that takes place to maintain hemoglobin concentration is what causes microcytosis.
Heme, which is formed of iron and protoporphyrin, and globin combine to form hemoglobin.
Microcytic anemia is caused by a reduction in any one of these components:
- Fe (iron)
- Protoporphyrin
- Globin
Examples of microcytic anemias include:
- Iron deficiency anemia
- Chronic disease-related anemia
- Sideroblastic anemia
- Thalassemia
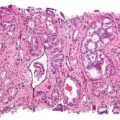
 Microcytic Anemias. Microcytosis is the presence of red cells that are smaller than normal. Normal adult red cell has a diameter of 7.2 µm. Microcytes are common seen in with hypochromia in iron-deficiency anaemia, thalassaemia trait, congenital sideroblastic anaemia and sometimes in anaemia of chronic diseases. Prof. Osaro Erhabor - Prof. Osaro Erhabor. Not altered. CC0
Microcytic Anemias. Microcytosis is the presence of red cells that are smaller than normal. Normal adult red cell has a diameter of 7.2 µm. Microcytes are common seen in with hypochromia in iron-deficiency anaemia, thalassaemia trait, congenital sideroblastic anaemia and sometimes in anaemia of chronic diseases. Prof. Osaro Erhabor - Prof. Osaro Erhabor. Not altered. CC0
Iron Deficiency Anemia
Insufficient levels of iron cause iron deficiency anemia.
Iron deficiency anemia is the most common type of anemia.
A third of the world’s population suffers from iron insufficiency.
Sources of iron include:
- Iron consumed in heme or meat-derived sources
- Iron consumed in non-heme or vegetable-derived sources
Iron is absorbed in the small intestine, mainly the duodenum.
The heme form of iron is more easily absorbed than non-heme sources of iron.
Enterocytes have both heme and non-heme divalent metal transporters (DMTl).
Transferrin carries iron in the blood to the liver and bone marrow macrophages for storage whereas ferroportin helps enterocytes move iron across the cell membrane into the circulation.
Because stored intracellular iron is linked to ferritin, it cannot undergo the Fenton reaction and produce free radicals.
Laboratory measurements of iron status include:
- Serum iron
- Transferrin
- Total iron binding capacity (TIBC)
- Percent saturation
- Ferritin
Iron in the blood is measured by serum iron.
Transferrin molecule binds iron.
Total iron-binding capacity (TIBC) is the capacity for transferrin to bind iron.
Percent saturation is the proportion of transferrin molecules that are iron-bound.
Ferritin reflects iron stores in macrophages and the liver.
Dietary deficiencies or blood loss are the usual causes of iron deficiency.
Infants may be iron deficient because breast milk is low in iron.
Typically, children’s diets are poor.
Peptic ulcer disease in men and menorrhagia or pregnancy in women in adults aged 20 to 50 are risk factors.
Iron deficiency is brought on in older persons who have colon polyps or cancer, which is common in America.
In the developing world, hookworm, also known as Ancylostoma duodenale and Necator americanus may be associated with iron deficiency.
Malnutrition, poor absorption, and gastrectomy are further risk factors for iron deficiency.
By preserving the Fe2+ form, which is easier to absorb than Fe3+, acid promotes iron absorption.
Stages of iron deficiency include:
- Storage iron is depleted
- Serum iron is depleted
- Normocytic anemia
Normocytic anemia occurs when the bone marrow produces fewer but normal-sized red blood cells (RBCs).
Microcytic hypochromic anemia occurs when the bone marrow produces smaller and fewer red blood cells (RBCs).
Clinical features of iron deficiency include:
- Anemia
- Koilonychia (pitted nails)
- Pica (eating dirt to get iron)
Iron deficiency anemia is treated by:
- Addressing the underlying cause
- Iron supplementation
 Iron Deficiency Anemia. Blood smear of a person with iron-deficiency anemia at 40X enhancement. Rjgalindo. Not altered. CC BY-SA 3.0
Iron Deficiency Anemia. Blood smear of a person with iron-deficiency anemia at 40X enhancement. Rjgalindo. Not altered. CC BY-SA 3.0



Plummer-Vinson Syndrome
Plummer-Vinson syndrome is a condition that is caused by chronic iron deficiency.
Plummer-Vinson syndrome is characterized by:
- Anemia
- Dysphagia
- Beefy-red tongue
- Esophageal web
- Atrophic glossitis
In Plummer-Vinson syndrome, web like growth of membranes in the esophagus make swallowing difficult.
Plummer-Vinson syndrome may increase the risk of esophageal cancer.
Anemia of Chronic Disease
Anemia of chronic disease is anemia due to diseases or disorders.
Examples of chronic diseases that can cause anemia of chronic disease include:
- Endocarditis
- Autoimmune disorders
- Cancer
Anemia of chronic disease is regarded as the anemia that people in hospitals experience the most frequently.
The liver produces acute phase reactants, such as hepcidin, as a result of chronic illness.
Hepcidin limits the transfer of iron from macrophages to erythroid precursors and inhibits the formation of erythropoietin (EPO), which sequesters iron in storage sites.
Hepcidin hides iron from bacteria so bacteria die.
In chronic diseases, the body thinks bacteria are around running amok, which. is why hepcidin is released, takes up the iron from circulation, and causes anemia of chronic disease.
Laboratory findings include:
- Ferritin increased
- Total iron binding capacity (TlBC) decreased
- Serum iron decreased
- % saturation decreased
- Free erythrocyte protoporphyrin (FEP) increased
The underlying reason must be addressed during treatment of anemia of chronic disease.
A particular group of patients, particularly those with cancer, benefit from exogenous erythropoietin (EPO).
 Anemia of Chronic Disease. Peripheral blood smear shows spherocytosis (black arrow), reticulocytosis (red arrow), and microcytic hypochromic red blood cells. Coombs negative autoimmune hemolytic anemia in Crohn's disease. Park BS, Park S, Jin K, Kim YM, Park KM, Lee JN, Kamesaki T, Kim YW - The American journal of case reports (2014). Not Altered. CC.
Anemia of Chronic Disease. Peripheral blood smear shows spherocytosis (black arrow), reticulocytosis (red arrow), and microcytic hypochromic red blood cells. Coombs negative autoimmune hemolytic anemia in Crohn's disease. Park BS, Park S, Jin K, Kim YM, Park KM, Lee JN, Kamesaki T, Kim YW - The American journal of case reports (2014). Not Altered. CC.
Sideroblastic Anemia
Sideroblastic anemia is a very rare blood disorder characterized by hemoglobin deficient red blood cells (RBCs).
Protoporphyrin is generated by a sequence of processes, and the protoporphyrin synthesis is deficient in sideroblastic anemia.
Using vitamin B6 as a cofactor, aminolevulinic acid synthetase (ALAS) transforms succinyl CoA to aminolevulinic acid (ALA) in a rate-limiting step.
Aminolevulinic acid dehydrogenase (ALAD), on the other hand, converts aminolevulinic acid (ALA) into porphobilinogen.
Porphobilinogen undergoes further processes that result in protoporphyrin.
Then, protoporphyrin is joined to iron by ferrochelatase to produce heme.
In the mitochondria, the last reaction takes place.
Heme is formed when iron enters the mitochondria after being transported to erythroid precursors.
In the absence of protoporphyrin, iron gets trapped in mitochondria.
A ring of iron-rich mitochondria surrounds the nucleus of erythroid precursors.
The word sideroblastic anemia comes from these cells, which are known as ringed sideroblasts.
Both congenital and acquired sideroblastic anemia exist.
Aminolevulinic acid synthetase (ALAS) deficiency is the most common cause of congenital sideroblastic anemia.
Acquired causes of sideroblastic anemia include:
- Lead poisoning
- Alcoholism
- B6 deficiency
- Isoniazide treatment (i.e. poor diet or tuberculosis (TB) patients)
Lead poisoning inhibits ferrochelatase and aminolevulinic acid dehydrogenase (ALAD).
Alcoholism results in mitochondrial poisoning.
Aminolevulinic acid synthetase (ALAS) requires vitamin B6 as a cofactor, and vitamin B6 deficiency is most frequently observed as a side effect of the isoniazid treatment for tuberculosis.
Laboratory findings of sideroblastic anemia include:
- Increased ferritin
- TIBC
- Increased serum iron
- Increased % saturation
 Sideroblastic Anemia. Sideroblast, as seen in sideroblastic anemia. Paulo Henrique Orlandi Mourao. Not altered. CC BY-SA 3.0
Sideroblastic Anemia. Sideroblast, as seen in sideroblastic anemia. Paulo Henrique Orlandi Mourao. Not altered. CC BY-SA 3.0

Thalassemia
Thalassemia is a red blood cell disorder caused by decreased synthesis of globin chains (which are needed to make hemoglobin).
Reduced production of the globin chains results in decreased hemoglobin which causes microcytic anemia.
Thalassemia patients and carriers of thalassemia genes are protected from Plasmodium Falciparum malaria due to a hereditary mutation.
There are two main groups of thalassemia:
- Alpha thalassemia
- Beta thalassemia
Alpha thalassemia
Alpha thalassemia is due to gene deletions.
Normal types of hemoglobin are HbF, HbA and HbA2.
Gene deletion is typically the cause of a-thalassemia.
On chromosome 16, four alpha genes are often found.
One gene deletion is not a big problem, patients will be asymptomatic.
Two gene deletions causes a mild anemia, with slightly increased red blood cell count.
Cis would be both on the same chromosome: worse than trans deletion.
Asians commonly experience cis deletions.
Trans deletion would be on different chromosomes: better than cis deletion.
Africans commonly experience trans deletions.
Three gene deletions cause a severe anemia that produces tetramers of beta resulting in hemoglobin H (HbH) that harm red blood cells (RBCs).
Electrophoresis reveals HbH.
Hydrops fetalis due to four genes removed.
RBC damage is caused by gamma chains forming tetramers (Hb Barts), which may be detected on an electrophoresis.
Beta thalassemia
Beta thalassemia is due to gene mutations.
People most commonly at risk for Beta thalassemia include:
- African
- Mediterranean
Chromosome 11 contain genes.
The chain is produced less (beta +) or not at all (beta 0) as a result of mutations.
The least severe form of Beta thalassemia is beta thalassemia minor (beta normal / beta +) disease.
Beta thalassemia minor is characterized by:
- Elevated red blood cell count (RBC)
- Asymptomatic
- Microcytic, hypochromic red blood cells (RBCs)
- Target cells
Hemoglobin electrophoresis of beta thalassemia minor shows slightly decreased HbA with increased HbA2 (5%, normal 5%) and HbF (2%, normal 2%).
The most severe form beta thalassemia is beta thalassemia major (beta 0 / beta 0).
Beta thalassemia major does not affect the fetus.
After birth, strong HbF (a2 y2) at birth provides momentary protection.
A few months after birth, the baby will finally become symptomatic.
In beta thalassemia major, tetramers cause red blood cells (RBCs) to cluster and get damaged, which impairs erythropoiesis and causes extravascular hemolysis or the spleen to remove circulating red blood cells (RBCs).
Massive erythroid hyperplasia that follows causes:
- The hematopoiesis to spread into the skull, where reactive bone development causes chipmunk faces
- Crewcut appearance of the skull due to hematopoiesis in the skull
- Extramedullary hematopoiesis with hepatosplenomegaly
Patients with beta thalassemia major are at risk due to parvovirus B19 infection of erythroid precursors.
Frequent need for chronic transfusions increases the risk of secondary hemochromatosis in beta thalassemia major.
Histology of beta thalassemia major will show a blood smear with microcytic, hypochromic red blood cells (RBCs) with target cells and nucleated red blood cells.
Electrophoresis of beta thalassemia major will show little to no HbA with elevated HbA2 and HbF.
 Thalassemia. Peripheral Blood film from a patient with Delta Beta Thalassemia. Prof. Osaro Erhabor. Not altered. CC BY-SA 4.0
Thalassemia. Peripheral Blood film from a patient with Delta Beta Thalassemia. Prof. Osaro Erhabor. Not altered. CC BY-SA 4.0