What Diseases are Caused by Trinucleotide-Repeats?
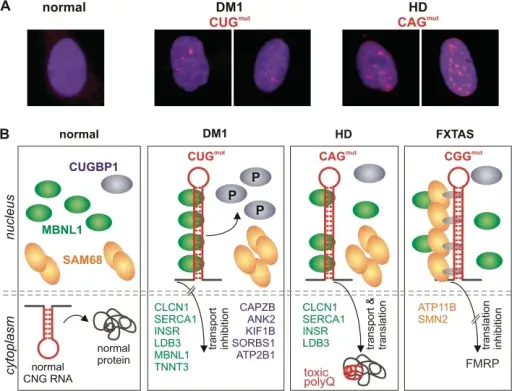
(A) Mutant CUG and CAG repeat RNAs form nuclear foci (red) in DM1 and HD human fibroblasts. RNA FISH was performed using fluorescently labelled repeat probes: CAG in DM1 cells and CTG in HD cells, normal cells were treated with either probe; (B) In normal cells, transcripts with short CNG repeats are exported from the nucleus and translated into functional proteins (first panel). Thus, the availability of nuclear splicing factors CUGBP1 (blue), MBNL1 (green) and SAM68 (orange) is not compromised. In DM1 cells (second panel), the CUGmut transcript is retained in the nucleus, where it sequesters MBNL1 and causes the abnormal phosphorylation of CUGBP1, upregulating its nuclear levels. As a result, the incorrect splicing of several MBNL1- and CUGBP1-dependent transcripts occurs (examples of misspliced transcripts are specified as green and blue, respectively). In HD cells (third panel), expanded CAG repeat RNA partially sequesters MBNL1, but does not change the level of CUGBP1, which results in splicing abnormalities affecting some MBNL1-dependent transcripts. The CAGmut transcript is effectively translated into toxic polyQ protein (polyQ tract is indicated by a red line). In FXTAS cells (fourth panel), the CGGmut transcript colocalizes in the nucleus with SAM68 and MBNL1, but this colocalization does not occur via direct RNA–protein interaction. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target: Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P - Nucleic acids research (2011). Not altered. CC.
X-linked spinal and bulbar muscular atrophy (SBMA)
Fragile X syndromes of mental retardation (FRAXA and FRAXE)
Myotonic dystrophy
Huntington’s disease
Spinocerebellar ataxia type 1 (SCA1)
Dentatorubral-pallidoluysian atrophy (DRPLA)
TRD
Mode of inheritance
Repeats involved
Examples of presentation
Dentatorubral-pallidoluysian atrophy
Autosomal dominant
CAG≥49 Chromosome 12
Symptom onset is variable and depends on trinucleotide repeat length: can present with ataxia, dementia, depression, anxiety, chorea, etc.
Problems of movement with cerebellar gait ataxia and action tremor.
Friedreich`s ataxia
Autosomal recessive
GAA≥70 Chromosome 9
Progressive ataxia, dysarthria, depression, etc.
Hunington’s disease
Autosomal dominant
CAG≥40 Chromosome 4
Chorea, dementia, depression.
Myotonic dystrophy
Autosomal dominant
CTG>50 Chromosome 19
Intellectual disability or psychiatric symptoms can occur but main presentation is usually with muscle weakness, etc.
Spinobulbar muscular atrophy
X-linked recessive
CAG≥38
Progressive muscles weakness and atrophy, fasciculation, etc.
Spinocerebellar ataxia
Autosomal recessive
Various, depending on the subtype
Variability in age of onset dependent on trinucleotide repeat length. Psychosis and dementia can occur with some subtypes, but progressive ataxia is the main presentation.