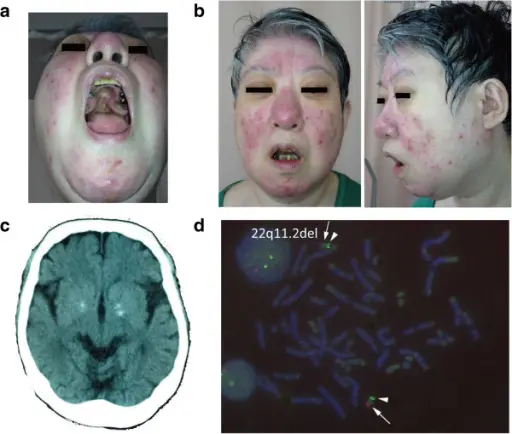
22q11.2 deletion syndrome is a syndrome caused by a microdeletion on the long arm of chromosome 22 resulting in abnormal development of several body systems.
What is the Pathology of 22q11.2 Deletion Syndrome?
The pathology of 22q11.2 deletion syndrome is:
-Etiology: The cause of 22q11.2 deletion syndrome is heterozygous deletion of part of the long arm (q) of chromosome 22, region 1, band 1, sub-band 2 (22q11.2).
-Genes involved: 1. Haploinsufficiency of the TBX1 gene (T-box transcription factor TBX1). Haploinsufficiency of the DGCR8 gene has been linked to improper regulation of the microRNA miR-338 and 22q11.2 deletion phenotypes. TANGO2 gene.
-Pathogenesis: 22q11.2 microdeletion causes to failure to develop 3rd and 4th pharyngeal pouches which leads to an absent thymus.
How does 22q11.2 Deletion Syndrome Present?
Patients with 22q11.2 deletion syndrome typically affects either male or female present at birth. The symptoms, features, and clinical findings associated with 22q11.2 deletion syndrome include congenital heart disease, (ventricular septal defect, tetralogy of Fallot, interrupted aortic arch, and truncus arteriosus), palatal abnormalities, and immune deficiency.
How is 22q11.2 Deletion Syndrome Diagnosed?
22q11.2 deletion syndrome diagnosis is established by identification of a heterozygous deletion at chromosome 22q11.2 on chromosomal microarray analysis or other genomic analyses.
How is 22q11.2 Deletion Syndrome Treated?
22q11.2 deletion syndrome is treated in order to certain individual features using standard treatments. No cure is known for DiGeorge syndrome.
What is the Prognosis of 22q11.2 Deletion Syndrome?
There is a wide range of symptoms and severity among people with 22q11.2 deletion syndrome. The long-term outlook for each person depends on the specific signs and symptoms each individual has.