Pheochromocytoma is a type of neuroendocrine, catecholamine-secreting tumor that grows from chromaffin cells that produce hormones needed for the body and are found in the adrenal glands.
What is the Pathology of Pheochromocytoma?
The pathology of pheochromocytoma is:
-Etiology: The cause of pheochromocytoma is excessive production of catecholamines.
-Genes involved: Familial Paraganglioma 1, 3, and 4, MEN-2A, MEN-2B, NF1, and VHL.
-Pathogenesis: The sequence of events that lead to pheochromocytoma is that it results from excessive catecholamine secretion by the tumor.
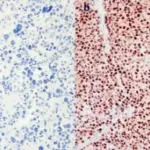
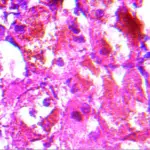
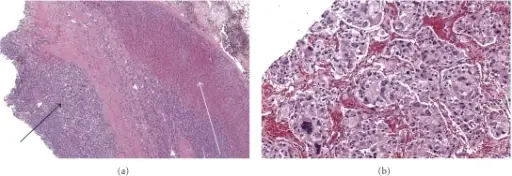
-Morphology: The morphology associated with pheochromocytoma shows a large mass of high signal intensity.
-Histology: The histology associated with pheochromocytoma shows increased cells in the adrenal medulla.
How does Pheochromocytoma Present?
Patients with pheochromocytoma typically are either male or female that present at the age range of 20-50. The symptoms, features, and clinical findings associated with pheochromocytoma include high blood pressure, headache, sweating, and symptoms of a panic attack, rapid heartbeat, tremors, paleness in the face, and shortness of breath.
How is Pheochromocytoma Diagnosed?
Pheochromocytoma is diagnosed with tests that measure levels of adrenaline, noradrenaline, or byproducts of those hormones.
How is Pheochromocytoma Treated?
Pheochromocytoma is treated with surgery to remove the tumor and patients usually take two drugs for seven to 10 days that help lower blood pressure before surgery.
What is the Prognosis of Pheochromocytoma?
The prognosis of pheochromocytoma is poor. After the surgery blood pressure usually returns to normal and even if all of the cancerous tissue isn’t removed, surgery might limit hormone production and provide some blood pressure control.