Inclusion body myositis is an inflammatory and degenerative disease that leads to painless muscle weakness.
What is the Pathology of Inclusion Body Myositis?
The pathology of Inclusion body myositis is:
-Etiology: The cause of inclusion body myositis is unknown. Viruses may be a factor the triggers the autoimmune activity of the disease.
-Pathogenesis: The sequence of events that lead to inclusion body myositis begins with an exogenous insult that triggers an autoimmune response.
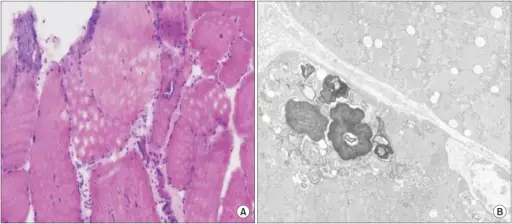
-Morphologic changes: The morphologic changes involved with inclusion body myositis is endomysial inflammation, atrophic fibers, eosinophilic cytoplasmic inclusions, and multiple myofibers with rimmed vacuoles line with granules.
How does Inclusion Body Myositis Present?
Patients with inclusion body myositis typically affects males with no racial predilection and present at age range of 56-60 years old. The symptoms, features, and clinical findings associated with inclusion body myositis include insidious and slowly progressive proximal leg and distal arm weakness with a hallmark of weakness and atrophy that commonly affects knee extensors and ankle dorsiflexors as well as wrist and finger flexors.
How is Inclusion Body Myositis Diagnosed?
Inclusion body myositis is diagnosed with laboratory tests on serum CK, ANA, as well as nerve conduction studies, needle examinations, and skeletal muscle MRI.
How is Inclusion Body Myositis Treated?
Inclusion body myositis is treated with corticosteroids and exercise therapy that can only result to transient and mild improvement in the course of the disease. No known effective treatment yet.
What is the Prognosis of Inclusion Body Myositis?
The prognosis of inclusion body myositis is good with life expectancy of patients not significantly altered, however most patients are bound to wheelchair 10-15 years after diagnosis.