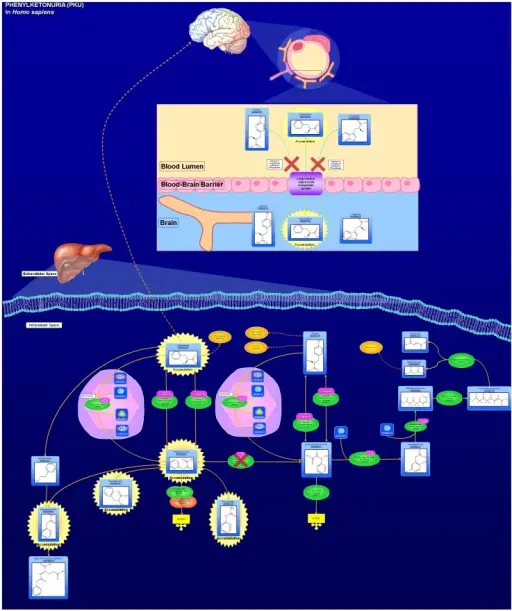
Phenylketonuria (PKU) is an autosomal recessive disorder caused by deficiency of the enzyme phenylalanine hydroxylase. Without phenylalanine hydroxylase, phenylalanine builds up in the patient, and may cause neurocognitive issues.
What is the Pathology of Phenylketonuria?
The pathology of phenylketonuria is:
–Etiology: The cause of phenylketonuria is caused by a defect in the gene that helps create the enzyme phenylalanine hydroxylase which is needed to break down phenylalanine. Phenylalanine builds up to high levels. Phenylalanine can build up to dangerous levels that cause neurocognitive issues.
–Genes involved: Mutations in the PAH gene.
–Pathogenesis: The sequence of events that lead to phenylketonuria are due to phenylalanine that cannot be metabolized by the body. Abnormally high levels of phenylalanine accumulate in the blood, which is toxic to the brain. If left untreated phenylketonuria may result in severe intellectual disability, brain function abnormalities, mood disorders, irregular motor functioning, and behavioral problems.
–Morphologic changes: The morphological changes involved with phenylketonuria are irreversible brain damage and marked intellectual disability beginning within the first few months of life.
How does Phenylketonuria Present?
Patients with phenylketonuria are typically baby males that usually seem healthy at birth. Signs of PKU begin to appear around six months of age. The symptoms, features, and clinical findings associated with phenylketonuria include neurological problems that may include seizures, eczema, microcephaly, delayed development, intellectual disability, and psychiatric disorders.
How is Phenylketonuria Diagnosed?
Phyenlketonuria is diagnosed by a blood test for the presence of the enzyme needed to break down phenylalanine.phenylketonuria is associated with attention deficit hyperactivity disorder, as well as physical symptoms such as a “musty” odor, eczema, and unusually light skin and hair coloration. Neurological problems such as seizures.
How is Phenylketonuria Treated?
Phenylketonuria is treated by diet modification to avoid foods contain phenylalanine.
What is the Prognosis of Phenylketonuria?
The prognosis of phenylketonuria is great if a phenylalanine restricted diet is followed, else it is can be poor. 2 is poor if not treated. If left untreated, PKU can cause brain damage or even death. However, if the condition is detected early and treatment is begun, individuals with phenylketonuria can lead healthy lives.