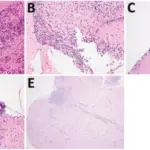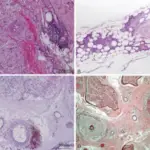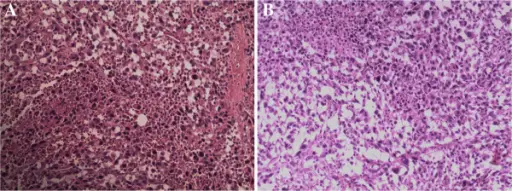
White blood cell pathology is a blood disorder in which the white blood cells are either abnormally low (leukopenia) or abnormally high (leukocytosis). There are two broad categories which include leukopenias and proliferative disorders (leukocytosis). The proliferation of white blood cells may be malignant or reactive.
What is Leukopenia?
Leukopenia is a condition where there is abnormally low number of white blood cells (leukocytes).
Types of leukopenia include:
- Agranulocytosis
- Neutropenia
What is Agranulocytosis?
Agranulocytosis is a condition that occurs when the bone marrow does not produce enough granulocytes or neutrophils (a type of white blood cell) resulting to an extremely low number of granulocytes in the blood. Agranulocytosis is a severe form of neutropenia with absolute neutrophil count (ANC) of less than 100 cells/microliter.
What is Neutropenia?
Neutropenia is a condition characterized by a lower- than- normal level of neutrophils (a type of white blood cell) in the blood with absolute neutrophil count of less than 500 cells/microliter.
What are White Blood Cell Proliferative Disorders?
White blood cell proliferative disorders are conditions where there is an increase in the number of white blood cells.
What are Reactive Proliferations of White Cells and Lymph Nodes?
Reactive proliferations of white cells and lymph nodes refer to events occurring in the white cells and lymph nodes in response to infection and inflammation resulting to an increase in it’s numbers.
Examples of reactive proliferations of white cells and lymph nodes include:
- Leukocytosis
- Lymphadenitis
- Acute nonspecific lymphadenitis
- Chronic nonspecific lymphadenitis
- Hemophagocytic lymphohistiocytosis
What is Leukocytosis?
Leukocytosis refers to an increase in the number of white blood cells (leukocytes). It is a common reaction to a variety of inflammatory states.
What is Acute Nonspecific Lymphadenitis?
Acute nonspecific lymphadenitis refers to acute inflammation of lymph nodes.
What is Chronic Nonspecific Lymphadenitis?
Chronic nonspecific lymphadenitis refers to chronic inflammation of lymph nodes.
What is Hemophagocytic Lymphohistiocytosis?
Hemophagocytic lymphohistiocytosis is a rare hematologic disorder characterized by an uncontrolled immune response with organ infiltration of lymphocytes and histiocytes, and organ damage caused by excessive production of pro-inflammatory cytokines.
What Are Neoplastic Proliferations of White Cells?
Neoplastic proliferations of white cells refer to an abnormal increase in growth and number of white cells. Common examples include myeloid neoplasms, lymphoid neoplasms, and langerhans cell histiocytosis.
What are Myeloid Neoplasms?
Myeloid neoplasms are tumors that arise from early hematopoietic progenitors.
Examples of myeloid neoplasms include:
- Acute myeloid leukemia
- Myelodysplastic syndromes
- Myeloproliferative disorders
What is Acute Myeloid Leukemia?
Acute myeloid leukemia is a tumor of hematopoietic progenitors caused by acquired oncogenic mutations that impede differentiation, leading to the accumulation of immature myeloid blasts in the marrow.
What is the Pathology of Acute Myeloid Leukemia?
The pathology of acute myeloid leukemia is:
-Etiology: The cause of acute myeloid leukemia is related to the replacement of the mature bone marrow cells with blasts which produces marrow failure leading to complications such as anemia, thrombocytopenia and neutropenia.
-Genes involved: TP53 or p53.
-Pathogenesis: The sequence of events that lead to acute myeloid leukemia involve four functional categories:
(1) Transcription factor mutations that interfere with normal myeloid differentiation
(2) Mutation of signaling proteins that result in constitutive activation of pro-growth survival pathways
(3) Mutations of genes that regulate or maintain the epigenome
(4) Mutations of TP53 or genes that regulate p53
-Histology: The histology associated with acute myeloid leukemia shows the presence of at least 20% myeloid blasts in the bone marrow. It may be present as myeloblasts or monoblasts. Myeloblasts have delicate nuclear chromatin, two to four nucleoli and more voluminous cytoplasm than lymphoblasts. The cytoplasm contain needle shaped, peroxidase-positive azurophilic granules called Auer rods. Monoblasts have folded or lobulated nuclei, lack Auer rods and are nonspecific esterase-positive.
How does Acute Myeloid Leukemia Present?
Acute myeloid leukemia may occur at any age but the incidence rises throughout life peaking after 60 years of age. The symptoms, features, and clinical findings associated with acute myeloid leukemia include complaints related to anemia, neutropenia and thrombocytopenia, most notably fatigue, fever and spontaneous mucosal and cutaneous bleeding. Cutaneous petechiae and ecchymoses, serosal hemorrhages into the linings of the body cavities and viscera and mucosal hemorrhages into the gingiva and urinary tract are common. Acute myeloid leukemia occasionally presents as a localized soft tissue mass called a myeloblastoma.
How is Acute Myeloid Leukemia Diagnosed?
Acute myeloid leukemia is diagnosed by physical examination done by a health care provider checking for signs of bruising, bleeding or infection, enlarged liver, spleen or lymph nodes. Blood tests (complete blood count and peripheral smear) and bone marrow biopsy are also used to diagnose acute myeloid leukemia.
How is Acute Myeloid Leukemia Treated?
Acute myeloid leukemia is treated with chemotherapy, monoclonal antibody therapy and stem cell or bone marrow transplant.
What is the Prognosis of Acute Myeloid Leukemia?
The prognosis of acute myeloid leukemia is poor.
What are Myelodysplastic Syndromes?
Myelodysplastic syndromes are a group of cancers in which immature blood cells in the bone marrow do not develop into mature blood cells. These cells stay within the bone marrow in an immature state. “Myelodysplastic” come from “myelo” which means blood cells and “dysplastic” which means abnormal cell development or growth.
Examples of myelodysplastic syndromes include:
- Acute myelogenous leukemia
What are Myeloproliferative Disorders?
Myeloproliferative disorders are a group of hematopoietic stem cell disorders characterized by abnormal proliferation of one or more hematologic cell lines (platelets, white blood cells and red blood cells).
Examples of myeloproliferative disorders include:
- Chronic myelogenous leukemia
- Polycythemia vera
- Essential thrombocythemia
- Primary myelofibrosis
- Systemic mastocytosis
- Chronic eosinophilic leukemia
What is Chronic Myelogenous leukemia?
Chronic myelogenous leukemia is a myeloproliferative disorder characterized by increased proliferation of granulocytic cell line and determined to have the Philadelphia chromosome/translocation t(9;22)(q34;q11.2).
What is the Pathology of Chronic Myelogenous leukemia?
The pathology of chronic myelogenous leukemia is:
-Etiology: The cause of chronic myelogenous leukemia is a rearrangement (translocation) of genetic material between chromosome 9 and chromosome 22 creating an abnormal fusion gene called BCR-ABL1.
-Genes involved: BCR- ABL1 gene; Philadelphia chromosome translocation.
-Pathogenesis: The sequence of events that lead to chronic myelogenous leukemia occurs when a pluripotent stem cell undergoes malignant transformation and clonal proliferation, leading to a striking overproduction of mature and immature granulocytes.
-Histology: The histology associated with chronic myelogenous leukemia shows an increased number of granulocytes and their immature precursors, including occasional blast cells.
How does Chronic Myelogenous Leukemia Present?
Patients with chronic myelogenous leukemia typically affects both sexes with a slight male predominance and present at age range of 54 to 64 years (median age at diagnosis). The symptoms, features, and clinical findings associated with chronic myelogenous leukemia include weakness, fatigue, weight loss, fever, bone pain and enlarged spleen.
How is Chronic Myelogenous Leukemia Diagnosed?
Chronic myelogenous leukemia is diagnosed by elevated granulocyte count on complete blood count and bone marrow aspiration/biopsy that shows the Philadelphia chromosome in cytogenetic or molecular studies such as fluorescent in situ hybridization (FISH) and reverse transcription polymerase chain reaction (RT-PCR) of the samples.
How is Chronic Myelogenous Leukemia Treated?
Chronic myelogenous leukemia is treated by tyrosine kinase inhibitors (eg, imatinib, nilotinib, dasatinib, bosutinib, ponatinib) and sometimes, allogeneic stem cell transplantation.
What is the Prognosis of Chronic Myelogenous Leukemia?
The prognosis of chronic myelogenous leukemia is fair.
What is Polycythemia Vera?
Polycythemia vera is a blood disorder that involves an overproduction of blood cells in the bone marrow (myeloproliferation). The overproduction of red blood cells is most dramatic, but the production of white blood cells and platelets are also elevated in most cases.
What is the Pathology of Polycythemia Vera?
The pathology of polycythemia vera is:
-Etiology: The cause of polycythemia vera is a malignant change in the genetic material (DNA) within a single cell of the bone marrow (clonal disorder).
-Genes involved: JAK2 gene.
-Pathogenesis: The sequence of events that lead to polycythemia vera involve a variation in the JAK2 gene that produces Janus kinase 2.
-Histology: The histology associated with polycythemia vera shows bone marrow hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size).
How does Polycythemia Vera Present?
Patients with polycythemia vera typically affects slightly more men than women and occurs most often in individuals more than 60 years old, but can affect individuals of any age. It is extremely rare in individuals under 20. The symptoms, features, and clinical findings associated with polycythemia vera include general nonspecific symptoms such as headaches, fatigue, weakness, dizziness, excessive sweating especially at night, and itchy skin that, in severe cases, may be worse after taking a shower or a warm bath. Eventually, the spleen becomes involved. It often becomes abnormally enlarged in individuals with polycythemia vera as it attempts to clear a greater number of blood cells than normal – a condition called splenomegaly.
How is Polycythemia Vera Diagnosed?
Polycythemia vera is diagnosed based upon a thorough clinical evaluation, detailed patient history, and various specialized tests. A complete blood count (CBC) may demonstrate elevated numbers of red blood cells and sometimes platelets and white blood cells. Hemoglobin and hematocrit levels are elevated in polycythemia vera despite abnormally low erythropoietin levels. In some cases, surgical removal and microscopic examination of bone marrow tissue (biopsy) may also be used to diagnose polycythemia vera.
How is Polycythemia Vera Treated?
Polycythemia vera is treated with phlebotomy and drugs such as hydroxyurea, busulfan, chlorambucil and radioactive phosphorus.
What is the Prognosis of Polycythemia Vera?
The prognosis of Polycythemia vera is good.
What is Essential Thrombocythemia?
Essential thrombocythemia is a blood disorder in which the bone marrow produces too many platelets.
What is the Pathology of Essential Thrombocythemia?
The pathology of essential thrombocythemia is:
-Etiology: The cause of essential thrombocythemia is acquiring (not inheriting) a somatic mutation in any of several genes, such as the JAK2 gene (most frequent), CALR gene, and rarely the MPL, THPO, or TET2 gene.
-Genes involved: JAK2 gene, CALR gene, MPL gene.
-Pathogenesis: The sequence of events that lead to essential thrombocythemia stem from the driver JAK2, CALR and MPL genes which when mutated cause myeloproliferative effects.
-Histology: The histology associated with essential thrombocythemia shows an increase in bone marrow cellularity with megarkaryocytic hyperplasia.
How does Essential Thrombocythemia Present?
Patients with essential thrombocythemia typically are women, middle aged or over age 60, though younger people can develop it too. The symptoms, features, and clinical findings associated with essential thrombocythemia include headache, dizziness, chest pain, fainting, temporary vision changes, numbness or tingling of the hands and feet, redness, throbbing and burning pain in the hands and feet depending on where the clot forms (often in the brain, hands and feet).
How is Essential Thrombocythemia Diagnosed?
Essential thrombocythemia is diagnosed by a platelet count of ≥450 × 109/L , bone marrow biopsy showing megakaryocyte lineage with increased numbers of enlarged, mature megakaryocytes with no significant increase in neutrophil granulopoiesis or erythropoiesis and, rarely, minor reticulin fibers, not meeting WHO criteria for CML, polycythemia vera (PV), primary myelofibrosis, myelodysplastic syndromes, or other myeloid neoplasms, and presence of JAK2, CALR or MPL mutation.
How is Essential Thrombocythemia Treated?
Essential thrombocythemia is treated by prescription drugs such as hydroxyurea, anagrelide, interferon alfa-2b or peginterferon alfa-2a perhaps along with low-dose aspirin to reduce platelet count. Plateletpheresis is also used during emergencies.
What is the Prognosis of Essential Thrombocythemia?
The prognosis of essential thrombocythemia is good. The life expectancy of patients with essential thrombocythemia is nearly that of the healthy population. Median survival is approximately 20 years. For patients younger than age 60 years, median survival is 33 years.
What is Primary Myelofibrosis?
Primary myelofibrosis is a rare bone marrow disorder characterized by abnormalities in blood cell production (hematopoiesis) and scarring (formation of fibrous tissue) within the bone marrow.
What is the Pathology of Primary Myelofibrosis?
The pathology of primary myelofibrosis is:
-Etiology: The cause of primary myelofibrosis is unknown (idiopathic). Approximately, 50 percent of people with primary myelofibrosis have a mutation of the JAK2 gene. Mutations in the CALR gene occur in approximately 20% of the patients.
-Genes involved: JAK2 gene, CALR gene, MPL gene.
-Pathogenesis: The sequence of events that lead to primary myelofibrosis results from neoplastic transformation of a multipotent bone marrow stem cell. These progeny cells stimulate bone marrow fibroblasts (which are not part of the neoplastic transformation) to secrete excessive collagen. –
-Histology: The histology associated with primary myelofibrosis shows hypocellular bone marrow with usually alternating cellular and hypocellular regions, atypical megakaryocytes, marked collagen or reticulin fibrosis.
How does Primary Myelofibrosis Present?
Patients with primary myelofibrosis typically are males, age range of 50 to 70 years old. The symptoms, features, and clinical findings associated with primary myelofibrosis include anemia, splenomegaly, or, in later stages, general malaise, weight loss, fever, or splenic infarction.
How is Primary Myelofibrosis Diagnosed?
Primary myelofibrosis is diagnosed by complete blood count and peripheral smear, bone marrow aspiration/biopsy and testing for JAK2, CALR and MPL mutations.
How is Primary Myelofibrosis Treated?
Primary myelofibrosis is treated with the therapy of choice, ruxolitinib (JAK pathway inhibitor). For patients with advanced disease, allogeneic stem cell transplantation may be beneficial.
What is the Prognosis of Primary Myelofibrosis?
The prognosis of primary myelofibrosis is poor. The median survival is 5 years from onset, but variation is wide; some patients have a rapidly progressing disorder, including development of acute myeloid leukemia, with short survival, but most have a more indolent course.
What is Systemic Mastocytosis?
Systemic mastocytosis is a blood disorder characterized by infiltration of clonally derived mast cells in different tissues, including bone marrow, the gastrointestinal (GI) tract, the liver, and the spleen.
What is the Pathology of Systemic Mastocytosis?
The pathology of systemic mastocytosis is:
-Etiology: The cause of systemic mastocytosis is a random change (somatic mutation) in the KIT gene
-Genes involved: KIT gene.
-Pathogenesis: The sequence of events that lead to systemic mastocytosis is the result of chronic and episodic discharge of mast cell mediators and excessive accumulation of mast cells in one or more tissues.
-Histology: The histology associated with systemic mastocytosis shows focal mast cell lesions in the bone marrow.
How does Systemic Mastocytosis Present?
Patients with systemic mastocytosis typically affects males and females equally, present mainly during adulthood with 55 years as median age at diagnosis. The symptoms, features, and clinical findings associated with systemic mastocytosis include flushing, itching or hives, abdominal pain, diarrhea, nausea or vomiting, anemia or bleeding disorders.
How is Systemic Mastocytosis Diagnosed?
Systemic mastocytosis is diagnosed by biopsies of tissues or organs affected revealing an abundance of mast cells, measuring mast cell mediators in blood and urine, blood counts, liver function studies and genetic tests.
How is Systemic Mastocytosis Treated?
Systemic mastocytosis is treated using antihistamines, steroids, epinephrine, mast cell stabilizers and other medications to relieve the symptoms. More aggressive forms may require interferon, immune modulators or chemotherapeutic agents.
What is the Prognosis of Systemic Mastocytosis?
The prognosis of systemic mastocytosis varies widely because systemic mastocytosis can range in severity. People who have more aggressive cases of mastocytosis may not survive more than a few years after diagnosis. People with less aggressive mastocytosis have a typical life expectancy.
What is Chronic Eosinophilic Leukemia?
Chronic eosinophilic leukemia is a chronic myeloproliferative diesease in which a clonal proliferation of eosinophilic precursor leads to increased eosinophils in blood, bone marrow, or peripheral tissues.
What is the Pathology of Chronic Eosinophilic Leukemia?
The pathology of chronic eosinophilic leukemia is:
-Etiology: The cause of chronic eosinophilic leukemia is unknown.
-Genes involved: Not linked to a specific gene or mutation.
-Pathogenesis: The sequence of events that lead to chronic eosinophilic leukemia.
-Histology: The histology associated with chronic eosinophilic leukemia shows striking eosinophilia in the peripheral blood and hypercellular bone marrow due to eosinophilic proliferation.
How does Chronic Eosinophilic Leukemia Present?
Patients with chronic eosinophilic leukemia typically involve males in the seventh decade of life. The symptoms, features, and clinical findings associated with chronic eosinophilic leukemia include fatigue, cough, dyspnea, myalgia, angioderma, rash, fever, and rhinitis.
How is Chronic Eosinophilic Leukemia Diagnosed?
Chronic eosinophilic leukemia is diagnosed by complete blood count (eosinophil count of at least 1.5 x 109/L ), bone marrow aspiration and biopsy showing hypercellularity with eosinophilic proliferation.
How is Chronic Eosinophilic Leukemia Treated?
Chronic eosinophilic leukemia is treated by using corticosteroids, interferon alfa, and chemotherapeutic drugs such as hydroxyurea, cyclophosphamide, and vincristine.
What is the Prognosis of Chronic Eosinophilic Leukemia?
The prognosis of chronic eosinophilic leukemia is poor. Survival is variable, with median survival of 22.2 months.
| Myeloproliferative Syndrome | Genetics | Key Histologic Features | Key Clinical Findings |
| Chronic myelogenous leukemia | Philadelphia chromosome/translocation | Increased granulocytes and immature precursors | Enlarged spleen, weakness, fatigue, fever, bone pain, weight loss |
| Polycythemia vera | Point mutation in JAK2 gene | Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size) | Splenomegaly, headaches, fatigue, weakness, dizziness, excessive sweating at night, itchy skin |
| Essential thrombocythemia | JAK2, CALR, MPL gene mutation | Increase in bone marrow cellularity with megarkaryocytic hyperplasia | Headache, dizziness, chest pain, fainting, temporary vision changes, numbness or tingling of the hands and feet, redness, throbbing and burning pain in the hands and feet |
| Primary myelofibrosis | JAK2, CALR, MPL gene mutation | Hypocellular bone marrow with marked reticulin or collagen fibrosis. | Anemia, splenomegaly, general malaise, weight loss, fever, splenic infarction. |
| Systemic mastocytosis | KIT gene somatic mutation | Focal mast cell lesions in the bone marrow. | Flushing, itching or hives, abdominal pain, diarrhea, nausea or vomiting, anemia or bleeding disorders |
| Chronic eosinophilic leukemia | Not linked to a specific gene or mutation | Striking eosinophilia in the peripheral blood and hypercellular bone marrow due to eosinophilic proliferation. | Fatigue, cough, dyspnea, myalgia, angioderma, rash, fever, rhinitis |
What are Lymphoid Neoplasms?
Lymphoid neoplasms are neoplasms that arise from the malignant transformation of normal lymphoid cells at various stages of differentiation.
Examples of lymphoid neoplasms include:
- Precursor B- and T-cell neoplasms
- Peripheral B-cell neoplasms
- Peripheral T-cell neoplasms
- NK-cell neoplasms
- Hodgkin lymphoma
What are Precursor B- Cell Neoplasms?
Precursor B- neoplasms are neoplasms involving lymphoblasts committed to the B-cell lineage.
An example includes B-cell acute lymphoblastic leukemia/lymphoma (B-ALL)
What is B-cell Acute Lymphoblastic Leukemia/lymphoma (B-ALL)?
B-cell acute lymphoblastic leukemia/lymphoma (B-ALL) is an aggressive (fast-growing) type of leukemia (blood cancer) in which too many B-cell lymphoblasts (immature white blood cells) are found in the bone marrow and blood.
What is the Pathology of B-cell Acute Lymphoblastic Leukemia/lymphoma (B-ALL)?
The pathology of b-cell acute lymphoblastic leukemia/lymphoma is:
-Etiology: The cause of b-cell acute lymphoblastic leukemia/lymphoma is unknown. Some things make this disease more likely, including exposure to high doses of X-rays and other forms of radiation, or cancer treatment with chemotherapy.
-Genes involved: PAX5, EBF1, IKZF1-3, lymphoid enhancer factor 1 genes.
-Pathogenesis: The sequence of events that lead to b-cell acute lymphoblastic leukemia/lymphoma involves a number of abnormal gene expressions (including TEL-AML1, BCR-ABL-1, RAS and PI3K) leading to dysregulated cell cycle.
-Histology: The histology associated with b-cell acute lymphoblastic leukemia/lymphoma shows blasts that tend to have FAB L3 features, with intensely staining cytoplasmic basophilia similar to that of erythroblasts.
How does B-cell Acute Lymphoblastic Leukemia/lymphoma (B-ALL) Present?
Patients with b-cell acute lymphoblastic leukemia/lymphoma typically are males (1.84:1 male to female ratio) present at age range of <15 years with a second peak of incidence around 40 years. The symptoms, features, and clinical findings associated with b-cell acute lymphoblastic leukemia/lymphoma include anemia, loss of appetite, fever, fatigue, bruising/bleeding, swollen or painful abdomen due to enlarged spleen and liver, bone and joint pain and recurring infections.
How is B-cell Acute Lymphoblastic Leukemia/lymphoma (B-ALL) Diagnosed?
B-cell acute lymphoblastic leukemia/lymphoma (B-ALL) is diagnosed by complete blood count, peripheral blood smear, bone marrow aspiration and biopsy, cerebrospinal fluid examination for cases with CNS involvement, immunophenotyping and chromosomal analysis.
How is B-cell Acute Lymphoblastic Leukemia/lymphoma (B-ALL) Treated?
B-cell acute lymphoblastic leukemia/lymphoma (B-ALL) is treated with chemotherapy, stem cell transplantation, targeted therapy and radiation therapy.
What is the Prognosis of B-cell Acute Lymphoblastic Leukemia/lymphoma (B-ALL)?
The prognosis of b-cell acute lymphoblastic leukemia/lymphoma is generally good in children but is not as favorable in adults. Five year survival for patients with B-cell ALL is greater than 90% in children, but only 40% to 50% in adults and elderly patients, respectively.
What are Precursor T-Cell Neoplasms?
Precursor T-cell neoplasms are neoplasms involving lymphoblasts committed to the T-cell lineage.
An example includes T-cell acute lymphoblastic leukemia/lymphoma (T-ALL).
What is the Pathology of T-cell Acute Lymphoblastic Leukemia/Lymphoma (T-ALL)?
The pathology of t-cell acute lymphoblastic leukemia/lymphoma is:
-Etiology: The cause of (t-cell acute lymphoblastic leukemia/lymphoma is unknown
-Genes involved: HOX 11, CFLAR, NOTCH 2 BTG3 genes.
-Pathogenesis: The sequence of events that lead to T-cell acute lymphoblastic leukemia/lymphoma involves transcriptional deregulation of oncogenes/oncosuppressors, NOTCH 1 signaling, cell cycle deregulation, kinase signaling pathway, epigenetic deregulation ribosomal dysfunction and altered expression of oncogenic miRNAs or long non coding RNA.
-Histology: The histology associated with t-cell acute lymphoblastic leukemia/lymphoma shows scant cytoplasm, delicate chromatin, indistinct nucleoli, convoluted nuclear membrane and grooves. Frequent mitotic figures and interspersed benign macrophages may be present. Complete architectural effacement or partial involvement with paracortical infiltrate with germinal center sparing of lymph nodes are seen.
How does T-cell Acute Lymphoblastic Leukemia/Lymphoma (T-ALL) Present?
Patients with t-cell acute lymphoblastic leukemia/lymphoma typically presents in older children with a male predominance. The symptoms, features, and clinical findings associated with (t-cell acute lymphoblastic leukemia/lymphoma are generally non-specific and are mostly due to bone marrow dysfunction. This include weakness, fatigue, shortness of breath, fever, malaise, night sweats, lymphadenopathy, pancytopenia, mucosal bleeding and purpura.
How is T-cell Acute Lymphoblastic Leukemia/Lymphoma (T-ALL) Diagnosed?
T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) is diagnosed mainly through biopsy of the bone marrow to determine the type of leukemia. Complete blood count and peripheral blood smears (Lymphoblasts of 20% or more of marrow cells) may be helpful.
How is T-cell Acute Lymphoblastic Leukemia/Lymphoma (T-ALL) Treated?
T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) is treated with chemotherapy along with steroids. Stem cell transplant is indicated only in younger patients that do not respond to chemotherapy.
What is the Prognosis of T-cell Acute Lymphoblastic Leukemia/Lymphoma (T-ALL)?
The prognosis of t-cell acute lymphoblastic leukemia/lymphoma is variable depending on the genes involved. HOX11 overexpression in adults indicate good prognosis while expressions of CFLAR, NOTCH 2, and BTG3 genes indicate poor prognosis.
What are Peripheral B-Cell Neoplasms?
Peripheral B-cell neoplasms are neoplasms involving mature B-cell lineage.
Examples of peripheral B-cell neoplasms include:
- Chronic lymphocytic leukemia/small lymphocytic lymphoma
- B-cell prolymphocytic leukemia
- Lymphoplasmacytic lymphoma
- Splenic marginal zone lymphoma
- Nodal marginal zone lymphomas
- Extranodal marginal zone lymphoma
- Mantle cell lymphoma
- Follicular lymphoma
- Marginal zone lymphoma
- Hairy cell leukemia
- Diffuse large B-cell lymphoma
- Burkitt lymphoma
- Plasma cell neoplasms and related conditions
What is Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma?
Chronic lymphocytic leukemia/small lymphocytic lymphoma is the most common leukemia of adults in the Western World. It is a type of leukemia involving lymphocytes that progresses more slowly than other types of leukemia. CLL and SLL differ only in the degree of peripheral blood lymphocytosis.
What is the Pathology of Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma?
The pathology of chronic lymphocytic leukemia/small lymphocytic lymphoma is:
-Etiology: The cause of chronic lymphocytic leukemia/small lymphocytic lymphoma is unknown.
-Genes involved: Deletions of 13q14.3, 11q, 17p and trisomy 12q.
-Pathogenesis: The sequence of events that lead to chronic lymphocytic leukemia/small lymphocytic lymphoma involve loss of tumor suppressor genes, and over expression of anti-apoptotic protein BCL2 that is observed in CLL/SLL.
-Histology: The histology associated with chronic lymphocytic leukemia/small lymphocytic lymphoma shows small lymphocytes with condensed chromatin and scant cytoplasm. A characteristic finding are Smudge cells, which are disrupted tumor cells. Larger activated lymphocytes that often gather in loose aggregates are referred to as proliferation centers, which are pathognomonic for CLL/SLL. These contain mitotically active cells.
How does Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma Present?
Patients with chronic lymphocytic leukemia/small lymphocytic lymphoma are typically diagnosed at a median age of 60 years and there is a 2:1 male predominance. The symptoms, features, and clinical findings associated with chronic lymphocytic leukemia/small lymphocytic lymphoma include easy fatigability, weight loss and anorexia. Most often patients are asymptomatic at diagnosis. Generalized lymphadenopathy and hepatosplenomegaly may be present.
How is Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma Diagnosed?
Chronic lymphocytic leukemia/small lymphocytic lymphoma is diagnosed variably. It may be through peripheral blood smear and a complete blood count or it may be through immunophenotyping. CLL/SLL has a distinctive immunophenotype. The tumor cells express the pan B-cell markers CD19, CD20, CD23 and CD5. Low level expression of surface Ig (usually IgM is also typical).
How is Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma Treated?
Chronic lymphocytic leukemia/small lymphocytic lymphoma is treated chemotherapy and immunotherapy with antibodies against proteins, particularly CD20.
What is the Prognosis of Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma?
The prognosis of chronic lymphocytic leukemia/small lymphocytic lymphoma is extremely variable and depend primarily on the clinical stage. Overall median survival is 4 to 6 years but is more than 10 years in individuals with minimal tumor burden at diagnosis.
What is B-cell Prolymphocytic Leukemia?
B-cell prolymphocytic leukemia is a very rare and typically aggressive malignancy (cancer) characterized by the out of control growth of B-cells (B-lymphocytes).
What is the Pathology of B-cell Prolymphocytic Leukemia?
The pathology of b-cell prolymphocytic leukemia is:
-Etiology: The cause of b-cell prolymphocytic leukemia is largely unknown.
-Genes involved: TP53 gene, MYC gene.
-Pathogenesis: The sequence of events that lead to b-cell prolymphocytic leukemia most of the time occurs as a transformation or evolution of a more slow-growing B-cell cancer, such as chronic lymphocytic leukemia.
-Histology: The histology associated with b-cell prolymphocytic leukemia shows large lymphoid cells (prolymphocytes) accounting for at least 55% of total circulating cells in the peripheral blood.
How does B-Cell Prolymphocytic Leukemia Present?
Patients with b-cell prolymphocytic leukemia typically are >60 years with a median age at diagnosis of 65-69 years. It is slightly more common in men than women. The symptoms, features, and clinical findings associated with b-cell prolymphocytic leukemia include high lymphocyte count, splenomegaly, B-symptoms (fevers, night sweats, weight loss), anemia and thrombocytopenia.
How is B-Cell Prolymphocytic Leukemia Diagnosed?
B-cell prolymphocytic leukemia is diagnosed by complete blood count, peripheral blood smear, bone marrow aspirate/biopsy, immunophenotyping by flow cytometry, cytogenetics either by karyotyping or FISH and molecular testing.
How is B-Cell Prolymphocytic Leukemia Treated?
B-cell prolymphocytic leukemia is treated by chemo-immunotherapy combinations used to treat chronic lymphocytic leukemia. Common regimens include fludarabine, cyclophosphamide, and rituximab or bendamustine and rituximab. In patients who achieve a remission following initial drug therapy, hematopoietic stem cell transplantation is a treatment option that may offer a potential cure.
What is the Prognosis of B-Cell Prolymphocytic Leukemia?
The prognosis of b-cell prolymphocytic leukemia is poor (median survival of 3 years). The presence of high-risk genetic mutations 17P/TP53 deletion translates into poor prognosis.
What is Lymphoplasmacytic Lymphoma?
Lymphoplasmacytic lymphoma is also known as Waldenstrom macroglobulinemia. It is a low-grade (slow-growing) B- cell lymphoproliferative neoplasm characterized by small lymphocytes and IgM monoclonal gammopathy.
What is the Pathology of Lymphoplasmacytic Lymphoma?
The pathology of lymphoplasmacytic lymphoma is:
-Etiology: The cause of lymphoplasmacytic lymphoma is poorly understood. However, the association of the disease with hepatitis C virus and autoimmune disorders has been documented.
-Genes involved: MYD88 gene (L265P mutation).
-Pathogenesis: The sequence of events that lead to lymphoplasmacytic lymphoma originate from cells at a late stage of B-cell differentiation. These cells derive from a B-cell arrest after somatic hypermutation in the germinal center and before terminal differentiation to a plasma cell.
-Histology: The histology associated with lymphoplasmacytic lymphoma shows infiltration of the bone marrow with small lymphocytes and IgM monoclonal gammopathy.
How does Lymphoplasmacytic Lymphoma Present?
Patients with lymphoplasmacytic lymphoma typically are elderly individuals in the seventh and eighth decade of life, with a slight male predominance. The symptoms, features, and clinical findings associated with lymphoplasmacytic lymphoma include B-related symptoms such as fever, night sweats, weight loss. Because of the frequent involvement of bone marrow, most lymphoplasmacytic lymphoma patients present with weakness and/or fatigue related to anemia. Some patients may present with the involvement of spleen, liver, and other extranodal sites, including skin, stomach, and bowel.
How is Lymphoplasmacytic Lymphoma Diagnosed?
Lymphoplasmacytic lymphoma is diagnosed by exclusion. A diagnosis should only be rendered after the exclusion of all other small B cell lymphomas. Diagnostic tests include complete blood count, peripheral blood smear, flow cytometry, protein electrophoresis and immunofixation.
How is Lymphoplasmacytic Lymphoma Treated?
Lymphoplasmacytic lymphoma is treated by close observation for asymptomatic patients. Symptomatic patients are managed by single-agent rituximab therapy without maintenance.
What is the Prognosis of Lymphoplasmacytic Lymphoma?
The prognosis of lymphoplasmacytic lymphoma is fair. The median survival of lymphoplasmacytic lymphoma patients is approximately five years. About 40% of patients survive for ten years or more.
What is Splenic Marginal Zone Lymphoma?
Splenic marginal zone lymphoma is a low-grade B-cell lymphoma that is unique to the spleen but that often secondarily involves the splenic hilar lymph nodes and usually involves the blood and bone marrow.
What is the Pathology of Splenic Marginal Zone Lymphoma?
The pathology of splenic marginal zone lymphoma is:
-Etiology: The cause of splenic marginal zone lymphoma is not known. It is more common in people who have been infected with hepatitis C virus and some autoimmune conditions.
-Genes involved: TP53 gene, NOTCH2 gene, KLF2 gene.
-Pathogenesis: The sequence of events that lead to splenic marginal zone lymphoma involves antigen or superantigen stimulation and molecular deregulation of different genes (including NOTCH2 and KLF2) involved in normal lymphocyte differentiation.
-Histology: The histology associated with splenic marginal zone lymphoma shows prominent expansion of the white pulp by small lymphocytes that often replace the normal germinal centers and efface the normal mantle zones.
How does Splenic Marginal Zone Lymphoma Present?
Patients with splenic marginal zone lymphoma typically have equal distribution between males and females, present at age range of 65-70 years. The median age at diagnosis is 65 years. The symptoms, features, and clinical findings associated with splenic marginal zone lymphoma include moderate to massive splenomegaly, autoimmune anemia, thrombocytopenia, and neutropenia which could be attributed to bone marrow infiltration and splenic sequestration. The majority of the patients show absolute lymphocytosis.
How is Splenic Marginal Zone Lymphoma Diagnosed?
Splenic marginal zone lymphoma is diagnosed by spleen histology; if that is not available, diagnosis requires integration of BM histology with cell morphology and immunophenotype in the blood and bone marrow.
How is Splenic Marginal Zone Lymphoma Treated?
Splenic marginal zone lymphoma is treated by splenectomy or splenic radiation, chemotherapy and targeted antibody therapy with rituximab.
What is the Prognosis of Splenic Marginal Zone Lymphoma?
The prognosis of splenic marginal zone lymphoma is fair . The median survival is approximately 8 to 10 years. The 10-year survival rate ranges from 67% to 95% with complete remission rate of 80%.
What is Nodal Marginal Zone Lymphoma?
Nodal marginal zone lymphoma is a primary lymph node disease with morphologic and immunophenotypic features similar to those of splenic marginal zone lymphoma and extranodal marginal zone lymphoma.
What is the Pathology of Nodal Marginal Zone Lymphoma?
The pathology of nodal marginal zone lymphomas is:
-Etiology: The cause of nodal marginal zone lymphomas is is not known. It is more common in people who have been infected with hepatitis C virus and some autoimmune conditions.
-Genes involved: Ig gene
-Pathogenesis: The sequence of events that lead to nodal marginal zone lymphomas involve many oncogenic mutations of NOTCH, NF-κB, B-cell receptor and toll-like receptor signaling in mature B-cells differentiation into the marginal zone B-cells.
-Histology: The histology associated with nodal marginal zone lymphomas shows perifollicular and interfollicular lymphoid infiltrate that surrounds variably preserved germinal centers; diffuse pattern of infiltration can also be seen.
How does Nodal Marginal Zone Lymphoma Present?
Patients with nodal marginal zone lymphoma typically are women more than men and present at age range of 60 years or older. The symptoms, features, and clinical findings associated with nodal marginal zone lymphomas include B symptoms, which are unexplained fever, drenching night sweats and unexplained weight loss, localized or generalized peripheral lymphadenopathy.
How is Nodal Marginal Zone Lymphoma Diagnosed?
Nodal marginal zone lymphoma is diagnosed by biopsy of affected lymph node.
How is Nodal Marginal Zone Lymphoma Treated?
Nodal marginal zone lymphoma is treated by watchful waiting, single chemotherapy (fludarabine or bendamustine), combination chemotherapy (cyclophosphamide, doxorubicin, vincristine and prednisone), targeted therapy (rituximab or ibritumomab), immunotherapy (interferon alpha) and radiation therapy.
What is the Prognosis of Nodal Marginal Zone Lymphoma?
The prognosis of nodal marginal zone lymphomas is less favorable than that of splenic marginal zone lymphoma and MALT lymphoma, with reported 5-year overall survival rates ranging between 55% and 89%.
What is Extranodal Marginal Zone Lymphoma?
Extranodal marginal zone lymphoma is a low-grade B-cell lymphoma that originate at virtually any extranodal site and arises in organs that normally lack lymphoid tissue (eg. stomach, intestine, thyroid, lung, and skin).
What is the Pathology of Extranodal Marginal Zone Lymphoma?
The pathology of extranodal marginal zone lymphoma is:
-Etiology: The cause of extranodal marginal zone lymphoma is chronic antigenic stimulation due to inflammation brought about by an infection.
-Genes involved: MALT1 gene, BCL10 gene
-Pathogenesis: The sequence of events that lead to extranodal marginal zone lymphoma has been shown to be associated with chronic immune reactions driven by bacterial, viral, or autoimmune stimuli.
-Histology: The histology associated with extranodal marginal zone lymphoma shows poorly defined follicular appearing areas that are composed of monocytoid B cells that feature enlarged nuclei.
How does Extranodal Marginal Zone Lymphoma Present?
Patients with extranodal marginal zone lymphoma typically have a slight female predominance, present at a wide age range of 21 – 92 years with mean age of 52-59 years. The symptoms, features, and clinical findings associated with extranodal marginal zone lymphoma include fever without an infection, night sweats, unexplained weight loss, skin rash, chest or abdominal pain, tiredness.
How is Extranodal Marginal Zone Lymphoma Diagnosed?
Extranodal marginal zone lymphoma is diagnosed by imaging tests such as X-rays, ultrasounds, CT scans and MRI scans.
How is Extranodal Marginal Zone Lymphoma Treated?
Extranodal marginal zone lymphoma is treated by antibiotic therapy if linked to an infection, corticosteroid therapy, chemotherapy, radiation therapy and surgery.
What is the Prognosis of Extranodal Marginal Zone Lymphoma?
The prognosis of extranodal marginal zone lymphoma is good with a 5-year survival rate of 88.7 percent.
What is Mantle Cell Lymphoma?
Mantle cell lymphoma is a lymphoproliferative disorder derived from a subset of naive pregerminal center cells localized in primary follicles or in the mantle region of secondary follicles.
What is the Pathology of Mantle Cell Lymphoma?
The pathology of mantle cell lymphoma is:
-Etiology: The cause of mantle cell lymphoma is not exactly known but approximately 85% of people with the condition have a genetic change, or mutation, in chromosomes 11 and 14.
-Genes involved: ATM, CCND1, TP53, MLL2, TRAF2 and NOTCH1 genes
-Pathogenesis: The sequence of events that lead to mantle cell lymphoma starts from the chromosomal translocation t(11;14) resulting in aberrant expression of cyclin D1. Secondary genetic events increase the oncogenic potential of cyclin D1 and frequently inactivate DNA damage response pathways. In combination these changes drive cell-cycle progression and give rise to pronounced genetic instability.
-Histology: The histology associated with mantle cell lymphoma shows expansion of the mantle zone that surrounds the lymph node germinal centers by small-to-medium atypical lymphocytes. These cells have irregular and indented nuclei, moderately coarse chromatin, and scant cytoplasm, resembling smaller cells of follicular lymphoma.
How does Mantle Cell Lymphoma Present?
Patients with mantle cell lymphoma typically have a male-to-female ratio of 3:1 and present at age range of 35-85 years, with a median of 68 years. The symptoms, features, and clinical findings associated with mantle cell lymphoma include fever, night sweats, weight loss, generalized lymphadenopathy, abdominal distention from hepatosplenomegaly, fatigue from anemia or bulky disease.
How is Mantle cell lymphoma Diagnosed?
Mantle cell lymphoma is diagnosed by lymph node biopsy and aspiration, bone marrow aspirate/biopsy, immunophenotyping and complete blood count (anemia, cytopenia and lymphocytosis). Body CT and PET scanning are important for initial staging.
How is Mantle Cell LymphomaTreated?
Mantle cell lymphoma is treated by combination chemotherapy typically in combination with the monoclonal antibody rituximab (Rituxan), as first-line treatment, followed by autologous stem cell transplantation.
What is the Prognosis of Mantle Cell Lymphoma?
The prognosis of mantle cell lymphoma is poor. Mantle cell lymphoma is not curable with conventional chemoimmunotherapy. Overall, the median survival is approximately 6 to 7 years.
What is Follicular Lymphoma?
Follicular lymphoma is the most common form of indolent non-hodgkin lymphoma in the United States affecting 15,000 to 20,000 individuals per year.
What is the Pathology of Follicular Lymphoma?
The pathology of follicular lymphoma is:
-Etiology: The cause of follicular lymphoma is unknown
-Genes involved: KMT2D gene
-Pathogenesis: The sequence of events that lead to follicular lymphoma involve chromosomal translocations involving BCL2. Its hallmark is a 14;18 translocation that juxtaposes IGH locus on chromosome 14 and the BCL2 locus on chromosome 18.
-Histology: The histology associated with follicular lymphoma shows a nodular and diffuse growth pattern in involved lymph nodes. Two cell types are present: 1) small cells with irregular or cleaved nuclear contours and scant cytoplasm, which are known as centrocytes; and 2) larger cells with open nuclear chromatin, several nucleoli and modest amounts of cytoplasm referred to as centroblasts.
How does Follicular Lymphoma Present?
Patients with follicular lymphoma typically present in middle ages and afflicts males and females equally. The symptoms, features, and clinical findings associated with follicular lymphoma include painless, generalized lymphadenopathy. It usually follows an indolent waxing and waning course.
How is Follicular Lymphoma Diagnosed?
Follicular Lymphoma is diagnosed through histology, specifically a biopsy of an affected lymph node. Immunophenotyping may also be done. Neoplastic cells resemble normal germinal centers CD19,CD20,CD10, surface Immunoglobulin and BCL6. Most cases are BCL2 positive.
How is Follicular Lymphoma treated?
Follicular lymphoma is treated with low-dose chemotherapy or immunotherapy (anti-CD20 antibody). It is also responsive to BTK inhibitors and BCL2 inhibitors.
What is the Prognosis of Follicular Lymphoma?
The prognosis of follicular lymphoma is fair with a median survival of 7 to 9 years. Histologic transformation into diffuse large B-cell lymphoma occurs in 30-50% of patients leading to a median survival of less than 1 year.
What is Marginal zone lymphoma?
Marginal zone lymphoma is a slow-growing type of B-cell non-Hodgkin lymphoma that begins forming in certain areas (marginal zones) of lymph tissue. There are three types of marginal zone lymphomas: the extranodal MZL (EMZL) of mucosa-associated lymphoid tissue (MALT or gastric GALT), the splenic MZL, and the nodal MZL.
What is the Pathology of Marginal zone lymphoma?
The pathology of marginal zone lymphoma is:
-Etiology: The cause of marginal zone lymphoma is unknown. There is a lot of evidence supporting the notion that antigen stimulation is important for the development and progression of marginal zone lymphoma.
-Genes involved: NOTCH gene, KLF2 gene, PTPRD gene.
-Pathogenesis: The sequence of events that lead to marginal zone lymphoma often associated with chronic antigenic stimulation of post-germinal center marginal zone B-lymphocytes.
-Histology: The histology associated with marginal zone lymphoma shows small to medium sized lymphocytes surrounding a reactive follicle. Plasmacytic differentiation is common.
How does Marginal zone lymphoma Present?
Patients with marginal zone lymphoma typically involve women than men present at age range of 60 years (average age at diagnosis). The symptoms, features, and clinical findings associated with marginal zone lymphoma include fever without an infection, night sweats, unexplained weight loss, skin rash, chest or abdominal pain and tiredness.
How is Marginal zone lymphoma Diagnosed?
Marginal zone lymphoma is diagnosed by X-rays, ultrasounds, CT scans, and MRI scans.
How is Marginal zone lymphoma Treated?
Marginal zone lymphoma is treated by chemotherapy to kill cancer cells, radiation to shrink tumors and surgery to remove tumors.
What is the Prognosis of Marginal zone lymphoma?
The prognosis of marginal zone lymphoma is good and most patients experience long survival.
What is Hairy Cell Leukemia?
Hairy cell leukemia is a rare indolent lymphoproliferative neoplasm of mature B cells.
What is the Pathology of Hairy Cell Leukemia?
The pathology of hairy cell leukemia is:
-Etiology: The cause of hairy cell leukemia is associated with point mutations in the serine/threonine kinase BRAF.
-Genes involved: BRAF serine/threonine kinase gene.
-Pathogenesis: The sequence of events that lead to hairy cell leukemia is associated with activating point mutations in the serine/threonine kinase BRAF which lies immediately downstream of RAS in the MPAK signaling cascade.
-Histology: The histology associated with hairy cell leukemia shows round, oblong or reniform nuclei and moderate amounts of pale blue cytoplasm with thread-like or bleb-like extensions. The number of circulating cells is highly variable. The marrow is involved by a diffuse interstitial infiltrate of cells with oblong or reniform nuclei, condensed chromatin and pale cytoplasm. In phase contrast microscopy, it shows tumor cells with fine hair-like cytoplasmic projections.
How does Hairy Cell Leukemia Present?
Patients with hairy cell leukemia typically involve middle-aged white males with a median age of 55 and a male to female ratio of 5:1. The symptoms, features, and clinical findings associated with hairy cell leukemia include largely from infiltration of the bone marrow, liver and spleen. Massive Splenomegaly is the most common and sometimes the only abnormal physical finding. Pancytopenia may be present and there is an increased incidence of atypical mycobacterial infections.
How is Hairy Cell Leukemia Diagnosed?
Hairy cell Leukemia is diagnosed mainly through peripheral blood and bone marrow morphology, flow cytometry, immunophenotyping, immunohistochemistry and molecular studies.
How is Hairy Cell Leukemia Treated?
Hairy cell Leukemia is treated with chemotherapeutic regimens which produce long lasting remissions. BRAF inhibitors may appear to produce excellent responses in tumors that have failed conventional chemotherapy.
What is the Prognosis of Hairy Cell Leukemia?
The prognosis of hairy cell leukemia is excellent due to its chronic relatively indolent course that responds well to current chemotherapy; 5 year event free survival rate after treatment is approximately 90%.
What is Diffuse Large B-Cell Lymphoma?
Diffuse Large B-cell Lymphoma is a neoplasm of large B lymphoid cells with nuclei at least twice the size of a lymphocyte.
What is the Pathology of Diffuse Large B-Cell Lymphoma?
The pathology of (disease in lower case) is:
-Etiology: The cause of diffuse large B-cell lymphoma is due to dysregulation of BCL6, overexpression of BCL2 and mutations in p300 and CREBP.
-Genes involved: BCL6, BCL2, p300 and CREBP genes.
-Pathogenesis: The sequence of events that lead to diffuse large B-cell lymphoma include dysregulation of BCL6, overexpression of BCL2 and mutations in p300 and CREBP.
-Histology: The histology associated with diffuse large B-cell lymphoma shows a relatively large cell size usually 4-5 times the diameter of a small lymphocyte and a diffuse pattern of growth. Tumor cells have a round or oval nucleus that appears vesicular due to margination of chromatin to the nuclear membrane. Cytoplasm is moderately abundant and may be pale or basophilic.
How does Diffuse Large B-Cell Lymphoma Present?
Patients with diffuse large B-cell lymphoma typically present with a slight male predominance and the median patient age is about 60 years. The symptoms, features, and clinical findings associated with diffuse large B-cell lymphoma typically include a rapidly enlarging mass at a nodal or extranodal site. The Waldeyer ring, oropharyngeal lymphoid tissue that includes the tonsils and adenoids, is involved most commonly. Involvement of the liver and spleen may take the form of a large destructive masses.
How is Diffuse Large B-Cell Lymphoma Diagnosed?
Diffuse Large B-cell Lymphoma is diagnosed mainly through biopsy of an enlarged lymph node and examining it through a microscope.
How is Diffuse Large B-Cell Lymphoma Treated?
Diffuse Large B-cell Lymphoma is treated with intensive combination chemotherapy. Adjuvant therapy with anti-CD20 improves both the initial response and overall outcome.
What is the Prognosis of Diffuse Large B-Cell Lymphoma?
The prognosis of diffuse large B-cell lymphoma is fair with remission rate of 60-80% and a cure rate of 40-50% when treated with intensive chemotherapy. DLBCLs are aggressive tumors that are rapidly fatal without treatment.
What is Burkitt Lymphoma?
Burkitt lymphoma is a highly aggressive non-hodgkin B cell lymphoma. They may occur in 3 categories: 1) African (endemic) Burkitt lymphoma, 2) sporadic (nonendemic) Burkitt lymphoma, and 3) a subset of aggressive lymphomas occurring in individuals infected with HIV.
What is the Pathology of Burkitt Lymphoma?
The pathology of burkitt lymphoma is:
-Etiology: The cause of burkitt lymphoma is primarily involving c-MYC rearrangement arising in germinal center derived B cells
-Genes involved: MYC gene
-Pathogenesis: The sequence of events that lead to burkitt lymphoma are associated with translocations of the MYC gene on chromosome 8 that lead to increased MYC protein levels. It was also seen that these tumors have an increased activity of the transcription factor TCF3 which leads to rapid growth.
-Histology: The histology associated with burkitt lymphoma shows tissues that are effaced by a diffuse infiltrate of intermediate-sized lymphoid cells 10 to 25 um in diameter with round or oval nuclei, coarse chromatin, several nucleoli and moderate amount of cytoplasm. The tumor exhibits a high mitotic index and contains numerous apoptotic cells, the nuclear remnants of which are phagocytosed by interspersed benign macrophages. These phagocytes have abundant clear cytoplasm creating a characteristic ‘starry sky’ pattern.
How does Burkitt Lymphoma Present?
Both endemic and sporadic Burkitt lymphomas are found mainly in children or young adults with a male predominance. Overall Burkitt lymphoma accounts for about 30% of childhood non-Hodgkin lymphomas in the United States. The symptoms, features, and clinical findings associated with burkitt lymphoma include: Endemic Burkitt lymphoma often presents as a mass involving the mandible and shows an unusual predilection for involvement of abdominal viscera (kidney, ovaries and adrenal glands). Sporadic Burkitt lymphoma most often appears as a mass involving the ileocecum and peritoneum.
How is Burkitt Lymphoma Diagnosed?
Burkitt Lymphoma is diagnosed mainly by karyotyping or FISH studies detecting C-MYC translocation. Elevated serum lactate dehydrogenase may raise its suspicion.
How is Burkitt Lymphoma Treated?
Burkitt Lymphoma is treated with intensive chemotherapy (R-CODOX-M / R-IVAC, (rituximab, cyclophosphamide, vincristine, doxorubicin, methotrexate, ifosfamide, etoposide, cytarabine ; or R-hyper-CVAD/ R-MA (rituximab, cyclophosphamide, vincristine, doxorubicin, dexamethasone, methotrexate, cytarabine).
What is the Prognosis of Burkitt Lymphoma?
The prognosis of burkitt lymphoma is fair with an event free survival in children of around 90% at 5 years and around 85% at 4 years in adults. Most children and young adults can be cured. The outcome is more guarded in older adults.
| Peripheral B-cell neoplasm | Genetics | Key Histologic Features | Key Clinical Findings |
| Chronic lymphocytic leukemia/small lymphocytic lymphoma | Deletions of 13q14.3, 11q, 17p and trisomy 12q | Small lymphocytes with condensed chromatin and scant cytoplasm. Smudge cells, which are disrupted tumor cells. Larger activated lymphocytes that often gather in loose aggregates are referred to as proliferation centers, which are pathognomonic for CLL/SLL | Easy fatigability, weight loss and anorexia, generalized lymphadenopathy, hepatosplenomegaly |
| B-cell prolymphocytic leukemia | TP53 gene, MYC gene | Large lymphoid cells (prolymphocytes) accounting for at least 55% of total circulating cells in the peripheral blood | High lymphocyte count, splenomegaly, B-symptoms (fevers, night sweats, weight loss), anemia and thrombocytopenia |
| Splenic marginal zone lymphoma | TP53 gene, NOTCH2 gene, KLF2 gene | Prominent expansion of the white pulp by small lymphocytes that often replace the normal germinal centers and efface the normal mantle zones | Massive splenomegaly, autoimmune anemia, thrombocytopenia, neutropenia, absolute lymphocytosis |
| Nodal marginal zone lymphoma | Ig gene | Perifollicular and interfollicular lymphoid infiltrate that surrounds variably preserved germinal centers; diffuse pattern of infiltration | Unexplained fever, drenching night sweats and unexplained weight loss, localized or generalized peripheral lymphadenopathy. |
| Extranodal marginal zone lymphoma | MALT1 gene, BCL10 gene | Poorly defined follicular appearing areas that are composed of monocytoid B cells that feature enlarged nuclei | Fever without an infection, night sweats, unexplained weight loss, skin rash, chest or abdominal pain, tiredness. |
| Mantle cell lymphoma | ATM, CCND1, TP53, MLL2, TRAF2 and NOTCH1 genes | Expansion of the mantle zone that surrounds the lymph node germinal centers by small-to-medium atypical lymphocytes | Fever, night sweats, weight loss, generalized lymphadenopathy, abdominal distention from hepatosplenomegaly, fatigue from anemia or bulky disease. |
| Follicular lymphoma | KMT2D gene | Nodular and diffuse growth pattern in involved lymph nodes. Two cell types are present: centrocytes and centroblasts | Painless, generalized lymphadenopathy |
| Marginal zone lymphoma | NOTCH gene, KLF2 gene, PTPRD gene | Small to medium sized lymphocytes surrounding a reactive follicle. Plasmacytic differentiation is common. | Fever without an infection, night sweats, unexplained weight loss, skin rash, chest or abdominal pain and tiredness |
| Hairy cell leukemia | BRAF serine/threonine kinase gene | Diffuse interstitial infiltrate of cells with round, oblong or reniform nuclei and moderate amounts of pale blue cytoplasm with thread-like or bleb-like extensions | Massive splenomegaly is the most common and sometimes the only abnormal physical finding |
| Diffuse large B-cell lymphoma | BCL6, BCL2, p300 and CREBP genes | Relatively large cell size usually 4-5 times the diameter of a small lymphocyte and a diffuse pattern of growth | Rapidly enlarging mass at a nodal or extranodal site. The Waldeyer ring, oropharyngeal lymphoid tissue that includes the tonsils and adenoids, are most commonly involved. |
| Burkitt lymphoma | MYC gene | Diffuse infiltrate of intermediate-sized lymphoid cells 10 to 25 um in diameter with round or oval nuclei, coarse chromatin, several nucleoli and moderate amount of cytoplasm. | Endemic Burkitt lymphoma often presents as a mass involving the mandible and shows an unusual predilection for involvement of abdominal viscera (kidney, ovaries and adrenal glands). Sporadic Burkitt lymphoma most often appears as a mass involving the ileocecum and peritoneum. |
What are Plasma cell neoplasms?
Plasma cell neoplasms are diseases in which the body makes too many plasma cells. Plasma cells develop from B lymphocytes (B cells), a type of white blood cell that is made in the bone marrow.
Examples of plasma cell neoplasms and related conditions include:
- Multiple myeloma
- MGUS
- Waldenstrom macroglobulinemia
- Heavy chain disease
- Primary or immunocyte-associated amyloidosis
- Plasmacytoma
What is Multiple Myeloma?
Multiple Myeloma is a plasma cell malignancy in which monoclonal plasma cells proliferate in bone marrow, resulting in an overabundance of monoclonal paraprotein (M protein), destruction of bone, and displacement of other hematopoietic cell lines.
What is the Pathology of Multiple Myeloma?
The pathology of multiple myeloma is:
-Etiology: The cause of multiple myeloma is not yet established. Roles have been suggested for a variety of factors, including genetic causes, environmental or occupational causes, MGUS, radiation, chronic inflammation, and infection.
-Genes involved: c-myc gene, N-Ras gene, K-ras gene.
-Pathogenesis: The sequence of events that lead to multiple myeloma is commonly preceded by MGUS, a premalignant condition that results when plasma cells undergo mutations that restore their capacity for proliferation.
-Histology: The histology associated with multiple myeloma shows mature myeloma cells usually indistinguishable from normal cells, with a round eccentric cartwheel nucleus without nucleoli and abundant basophilic cytoplasm. Immature myeloma cells have an irregular nucleus with more dispersed chromatin, a higher N/C ratio, and usually prominent nucleoli.
How does Multiple Myeloma Present?
Patients with multiple myeloma typically affects men slightly more than women,) present at age over 60 years. The average age at diagnosis is 70 years. The symptoms, features, and clinical findings associated with multiple myeloma include bone pain, pathologic fractures, spinal cord compression, weakness, malaise, anemia, bleeding, hypercalcemia, kidney failure, neuropathies.
How is Multiple Myeloma Diagnosed?
Multiple myeloma is diagnosed by complete blood count and differential, erythrocyte sedimentation rate, 24-hour urine collection for quantification of the Bence Jones protein, protein, and creatinine clearance; proteinuria greater than 1 g/24 hr is a major criterion, C-reactive protein, very high M protein.
How is Multiple Myeloma Treated?
Multiple myeloma is treated by chemotherapy and immunosuppression, peripheral blood or bone marrow stem cell transplantation.
What is the Prognosis of Multiple Myeloma?
The prognosis of multiple myeloma is fair, with survival ranging from 1 year to more than 10 years. Median survival in unselected patients with MM is 3 years. The 5-year relative survival rate is 46.6%.
What is MGUS?
MGUS is monoclonal gammopathy of unknown significance. It involves the production of M-protein by noncancerous plasma cells.
What is the Pathology of MGUS?
The pathology of mgus is:
-Etiology: The cause of mgus is not known. Infection, immune system problems, and the environment may play a role. –
-Genes involved: DCC gene.
-Pathogenesis: The sequence of events that lead to mgus come from clonal plasma cells in the bone marrow. These cells harbor somatic hypermutation of the variable regions and are class-switched.
-Histology: The histology associated with mgus shows bone marrow <10% plasma cells.
How does MGUS Present?
Patients with mgus typically affects men more than women and present at age range of 50-75 years. The symptoms, features, and clinical findings associated with mgus include bone pain, fatigue or weakness, unintentional weight loss, fever or night sweats, peripheral neuropathy, enhanced bone loss and fractures.
How is MGUS Diagnosed?
MGUS is diagnosed by blood and urine screening of abnormal proteins with a lab test called electrophoresis and immunofixation.
How is MGUS Treated?
No treatment is recommended for patients with MGUS. Long-term follow-up is generally advised.
What is the Prognosis of MGUS?
The prognosis of mgus is good. The annual risk of progression to multiple myeloma (MM), Waldenström macroglobulinemia (WM), amyloidosis (AL), or other lymphoproliferative disorders is approximately 1%. However, the mode and risk of progression vary between IgM MGUS and those with non-IgM MGUS.
What is Waldenstrom Macroglobulinemia?
Waldenstrom macroglobulinemia is a type of non-Hodgkin lymphoma (NHL). The cancer cells make large amounts of an abnormal protein (called a macroglobulin). Another name for WM is lymphoplasmacytic lymphoma.
What is the Pathology of Waldenstrom Macroglobulinemia?
The pathology of waldenstrom macroglobulinemia is:
-Etiology: The cause of waldenstrom macroglobulinemia is not definitely known. Environmental familial, genetic, and viral factors have been reported, –
-Genes involved: MYD88 gene
-Pathogenesis: The sequence of events that lead to waldenstrom macroglobulinemia is preceded by an IgM monoclonal gammopathy of undetermined significance (MGUS) in most, perhaps all, patients.
-Histology: The histology associated with waldenstrom macroglobulinemia shows the bone marrow containing variable numbers of pleomorphic lymphoid cells. Dutcher bodies may be seen as intracytoplasmic inclusions positive for periodic acid Schiff. Mast cell hyperplasia is common and may stimulate tumor cell proliferation and monoclonal IgM secretion. Blood smears show striking rouleau formation and occasional to frequent plasmacytoid lymphocytes which bear surface monoclonal IgM, usually κ, detected by flow cytometry.
How does Waldenstrom Macroglobulinemia Present?
Patients with waldenstrom macroglobulinemia typically involve males more than females and present at median age of 63 years. The symptoms, features, and clinical findings associated with waldenstrom macroglobulinemia include insidious weakness and mucous membrane bleeding. Some patients have infections, dyspnea, and congestive heart failure. Physical examination may detect pallor, purpura, lymphadenopathy, hepatosplenomegaly (20%) and engorged retinal veins.
How is Waldenstrom Macroglobulinemia Diagnosed?
Waldenstrom macroglobulinemia is diagnosed by blood test showing an increase in IgM on protein electrophoresis associated with ≥10% clonal lymphoplasmacytic cells in bone marrow aspiration/biopsy.
How is Waldenstrom macroglobulinemia Treated?
Waldenstrom macroglobulinemia is treated by combination therapy with rituximab and bendamustine.
What is the Prognosis of Waldenstrom Macroglobulinemia?
The prognosis of Waldenstrom macroglobulinemia is good. The five-year survival rate of Waldenstrom’s macroglobulinemia is about 78 percent. Recent studies suggest median survival rates closer to 14-16 years after diagnosis. This, plus the fact that people with WM tend to be older when diagnosed, puts their survival rates closer to those expected for the general population.
What is Heavy Chain Disease?
Heavy chain disease is a B-cell proliferative disorder characterized by production of abnormal, structurally incomplete, immunoglobulin heavy chains without the corresponding light chains.
What is the Pathology of Heavy Chain Disease?
The pathology of heavy chain disease is:
-Etiology: The cause of heavy chain disease is unknown.
-Genes involved: heavy chain gene
-Pathogenesis: The sequence of events that lead to heavy chain disease appear to result from structural genetic mutations in heavy chain proteins with internal deletions.
-Histology: The histology associated with heavy chain disease shows proliferation of B cells and extensive lymphoplasmacytic or plasmacytic infiltrate.
How does Heavy Chain Disease Present?
Patients with heavy chain disease typically are predominantly males, present at median age 58 years at diagnosis. The symptoms, features, and clinical findings associated with heavy chain disease include fever, mild anemia, difficulty swallowing (dysphagia), recurrent upper respiratory tract infection, lymphadenopathy, enlarged liver and spleen.
How is Heavy Chain Disease Diagnosed?
Heavy chain disease is diagnosed by documentation of a deleted immunoglobulin heavy chain without a bound light chain in the serum or urine. Bone marrow or lymph node biopsy done if other tests are not diagnostic, reveals variable histopathology.
How is Heavy Chain Disease Treated?
Heavy chain disease is treated by chemotherapy using alkylating agents and corticosteroids.
What is the Prognosis of Heavy Chain Disease
The prognosis of heavy chain disease is variable. The median survival with aggressive disease is about 1 year.
What is Primary or Immunocyte-Associated Amyloidosis?
Primary or immunocyte-associated amyloidosis is a type of amyloidosis that is immunocyte-derived and associated with elevated immunoglobulin free light chains in serum produced by monoclonal plasma cells.
What is the Pathology of Primary or Immunocyte-Associated Amyloidosis?
The pathology of primary or immunocyte-associated amyloidosis is:
-Etiology: The cause of primary or immunocyte-associated amyloidosis is caused by acquired overexpression of clonal immunoglobulin light chains.
-Genes involved: CCND1 (cyclin D1) gene.
-Pathogenesis: The sequence of events that lead to primary or immunocyte-associated amyloidosis come from plasma cells that produce extra pieces of antibodies called “light chains” These light chains circulate in the bloodstream, and can deposit in organs throughout the body, causing organ damage.
-Histology: The histology associated with primary or immunocyte-associated amyloidosis shows amyloid deposition in tissues and organs affected.
How does Primary or Immunocyte-Associated Amyloidosis Present?
Patients with primary or immunocyte-associated amyloidosis typically are thought to be equal in males and females. However, about 60% of patients referred to amyloid centers are males. They present at age range of 50-65 years. The symptoms, features, and clinical findings associated with primary or immunocyte-associated amyloidosis include changes in skin color, severe fatigue, feeling of fullness, anemia, joint pain, shortness of breath, tingling and numbness in hands and feet, severe weakness, sudden weight loss.
How is Primary or Immunocyte-Associated Amyloidosis Diagnosed?
Primary or immunocyte-associated amyloidosis is diagnosed by biopsy of affected tissue; the amyloidogenic protein is typed using a variety of immunohistologic and biochemical techniques.
How is Primary or Immunocyte-Associated Amyloidosis Treated?
Primary or immunocyte-associated amyloidosis is treated by standard-dose combination chemotherapy with steroids and alkylating agents. Myeloablative chemotherapy with melphalan and autologous stem cell rescue appears to offer survival benefit. Newer agents such as bortezomib and lenalidomide have shown promising activity and are being evaluated as part of combination regimens in clinical trials.
What is the Prognosis of Primary or Immunocyte-Associated Amyloidosis?
The prognosis of primary or immunocyte-associated amyloidosis is generally poor especially when the heart and kidneys are affected.
What is a Plasmacytoma?
Plasmacytoma is a tumor of plasma cells of bony or soft tissue and can occur anywhere in the body without evidence of systemic disease.
What is the Pathology of Plasmacytoma?
The pathology of plasmacytoma is:
-Etiology: The cause of plasmacytoma is unknown. It is suggested that it may be caused by inhalation of chemicals, chronic stimulation, an overdose of irradiation, viral infection, and genetic disorders in the reticuloendothelial system.
-Genes involved: CCND1 gene.
-Pathogenesis: The sequence of events that lead to plasmacytoma show that losses of chromosome 13 arm 1p and 14q and gains in arm 19,1q and 9q play an essential part in tumorigenesis of plasmacytoma.
-Histology: The histology associated with plasmacytoma shows monoclonal cell infiltrates in one or more lytic bone lesions or extramedullary tissue.
How does Plasmacytoma Present?
Patients with plasmacytoma typically are males more than females and present at age range of 55 to 60 years. The symptoms, features, and clinical findings associated with plasmacytoma include pain in the affected bone, back pain and other consequences of the bone lesion.
How is Plasmacytoma Diagnosed?
Plasmacytoma is diagnosed by lesional biopsy or fine needle aspiration techniques.
How is Plasmacytoma Treated?
Plasmacytoma is treated with surgery, radiotherapy and chemotherapy.
What is the Prognosis of Plasmacytoma?
The prognosis of plasmacytoma is poor. The causes of poor prognosis include local recurrence, incomplete resection with functional damage, local lymph node metastasis, progression to multiple myeloma, or new bone lesions formation with multiple myeloma.
| Plasma cell Neoplasms | Genetics | Key Histologic Findings | Key Clinical Findings |
| Multiple myeloma | c-myc gene, N-Ras gene, K-ras gene | Mature myeloma cells usually indistinguishable from normal cells, with a round eccentric cartwheel nucleus without nucleoli, abundant basophilic cytoplasm. Immature myeloma cells have an irregular nucleus with more dispersed chromatin, a higher N/C ratio, and usually prominent nucleoli. | Bone pain, pathologic fractures, spinal cord compression, weakness, malaise, anemia, bleeding, hypercalcemia, kidney failure, and neuropathies. |
| MGUS | DCC gene | Bone marrow <10% plasma cells | Bone pain, fatigue or weakness, unintentional weight loss, fever or night sweats, peripheral neuropathy, enhanced bone loss and fractures. |
| Waldenstrom macroglobulinemia | MYD88 gene | Bone marrow containing variable numbers of pleomorphic lymphoid cells. Dutcher bodies may be seen as intracytoplasmic inclusions positive for periodic acid Schiff. Mast cell hyperplasia is common and may stimulate tumor cell proliferation and monoclonal IgM secretion. | Insidious weakness and mucous membrane bleeding. Some patients have infections, dyspnea, and congestive heart failure. Physical examination may detect pallor, purpura, lymphadenopathy, hepatosplenomegaly (20%) and engorged retinal veins. |
| Heavy chain disease | Heavy chain gene | Proliferation of B cells and extensive lymphoplasmacytic or plasmacytic infiltrate. | Fever, mild anemia, difficulty swallowing (dysphagia), recurrent upper respiratory tract infection, lymphadenopathy, enlarged liver and spleen. |
| Primary or immunocyte-associated amyloidosis | CCND1 (cyclin D1) gene | Amyloid deposition in tissues and organs affected. | Changes in skin color, severe fatigue, feeling of fullness, anemia, joint pain, shortness of breath, tingling and numbness in hands and feet, severe weakness, sudden weight loss. |
| Plasmacytoma | CCND1 gene | Monoclonal cell infiltrates in one or more lytic bone lesions or extramedullary tissue. | Pain in the affected bone, back pain and other consequences of the bone lesion. |
What are Peripheral T-Cell Neoplasms?
Peripheral T-Cell Neoplasms are a group of aggressive lymphomas that develop from mature-stage white blood cells, T cells and NK cells.
Examples of Peripheral T-Cell Neoplasms include:
- T-cell prolymphocytic leukemia
- Large granular lymphocytic leukemia
- Mycosis fungoides
- Sézary syndrome
- Anaplastic large-cell lymphoma
- Angioimmunoblastic T-cell lymphoma
- Enteropathy-associated T-cell lymphoma
- Panniculitis-like T-cell lymphoma
- Hepatosplenic γδT-cell lymphoma
- Adult T-cell leukemia/lymphoma
- Peripheral T-cell lymphoma, unspecified
What is T-cell prolymphocytic leukemia?
T-cell prolymphocytic leukemia is an aggressive T cell leukemia comprised of small to medium sized mature T cells with high white blood cell count and widespread organ involvement.
What is the Pathology of T-cell prolymphocytic Leukemia?
The pathology of t-cell prolymphocytic leukemia is:
-Etiology: The cause of t-cell prolymphocytic leukemia is unknown but studies suggest patients with ataxia-telangiectasia are at higher risk.
-Genes involved: AKT.
-Pathogenesis: The sequence of events that lead to t-cell prolymphocytic leukemia include combination of overexpression of TCL1 family of proteins (this stimulate AKT/protein kinase B driven proliferation) and functional deficit of ATM protein
-Histology: The histology associated with t-cell prolymphocytic leukemia shows perivascular and diffuse tissue infiltrates of uniform small to medium sized lymphocytes.
How does T-cell prolymphocytic Leukemia Present?
Patients with t-cell prolymphocytic leukemia typically presents in the adults and elderly >30 years with a median age of 65 years with no gender predilection. The symptoms, features, and clinical findings associated with t-cell prolymphocytic leukemia include prominent constitutional symptoms of weight loss, fatigue, night sweats, fever and body malaise, cytopenias with bone marrow involvement, hepatosplenomegaly, lymphadenopathy and skin and mucosal lesions. Patients with high tumor burden have high WBC count.
How is T-cell prolymphocytic Leukemia Diagnosed?
T-cell prolymphocytic Leukemia is diagnosed by bone marrow or solid tissue biopsy of the involved organ (lymph node, spleen, liver, skin) and analyzing its morphology. Peripheral blood morphology, flow cytometry and variable fluorescent in situ hybridization may help in its diagnosis.
How is T-cell prolymphocytic Leukemia Treated?
T-cell prolymphocytic Leukemia is treated by alemtuzumab, an anti-CD52. BCL2, JAK3 or HDAC inhibitors were used as experimental therapies.
What is the Prognosis of T-cell Prolymphocytic Leukemia?
The prognosis of t-cell prolymphocytic leukemia is poor with a median survival of 1-2 years on patients with active disease.
What is Large Granular Lymphocytic Leukemia?
Large granular lymphocytic leukemia is a chronic T cell lymphoproliferative disorder characterized by a clonal proliferation of mature cytotoxic T cells.
What is the Pathology of Large Granular Lymphocytic Leukemia?
The pathology of large granular lymphocytic leukemia is:
-Etiology: The cause of large granular lymphocytic leukemia is chronic antigen exposure from autoimmune disorders such as rheumatoid arthritis and Sjogren syndrome.
-Genes involved: None.
-Pathogenesis: The sequence of events that lead to large granular lymphocytic leukemia involve acquired mutations in the transcription factor STAT 3, which functions downstream of cytokine receptor. This result in cytokine-independent activation of STAT 3 which favors proliferation.
-Histology: The histology associated with large granular lymphocytic leukemia shows large lymphocytes with abundant blue cytoplas and a few coarse azurophilic granules. The marrow contains sparse interstitial lymphocytic infiltrate.
How does Large Granular Lymphocytic Leukemia Present?
Patients with large granular lymphocytic leukemia typically are elderly individuals with a median age range of 60 years and no gender predilection.
The symptoms, features, and clinical findings associated with large granular lymphocytic leukemia include mild to moderate lymphocytosis, splenomegaly, neutropenia, anemia, cytopenias and history of autoimmune disorders (rheumatoid arthritis). Neutropenia may be accompanied with a decrease in late myeloid forms.
How is Large Granular Lymphocytic Leukemia Diagnosed?
Large Granular Lymphocytic Leukemia is diagnosed by peripheral blood smears showing persistent increase in the number of peripheral blood large granular lymphocytes. Molecular or flow cytometry may be helpful.
How is Large Granular Lymphocytic Leukemia Treated?
Large Granular Lymphocytic Leukemia is treated with immunosuppressive therapy of methotrexate, cyclophosphamide as initial agents. Low dose chemotherapy may be used. No standard therapy is established.
What is the Prognosis of Large Granular Lymphocytic Leukemia?
The prognosis of large granular lymphocytic leukemia is variable due to its indolent course and being largely dependent on the severity of the cytopenias and responsiveness to treatment.
What is Mycosis Fungoides?
Mycosis fungoides is a lymphoma of skin-homing CD4+ T-helper cells presenting in the skin.
What is the Pathology of Mycosis Fungoides?
The pathology of mycosis fungoides is:
-Etiology: The cause of mycosis fungoides is unknown
-Genes involved: None.
-Pathogenesis: The sequence of events that lead to mycosis fungoides involve proliferating cells that are clonal populations of CD4+ T-helper cells that home to the skin due to expression of cutaneous lymphocyte antigen. The neoplastic cells have clonal T-cell receptor gene rearrangements and may express aberrant combinations of T-cell surface antigens.
-Histology: The histology associated with mycosis fungoides shows the presence of atypical cells that form band-like aggregates with the superficial dermis and invade the epidermis as single cells and small clusters. These cells have markedly infolded nuclear membranes, imparting a convoluted or cerebriform contour.
How does Mycosis Fungoides Present?
Patients with mycosis fungoides typically affect patients older than age 40 but may occur at any age and is frequently seen in men than women. The symptoms, features, and clinical findings associated with mycosis fungoides include lesions involving truncal areas and include scaly, red brown patches; raised scaling plaques and fungating nodules. Lesions may affect numerous body surfaces including the trunk, extremities, face and scalp.
How is Mycosis Fungoides Diagnosed?
Mycosis fungoides is diagnosed mainly through exclusion correlating clinical, histologic and molecular data. Smear, flow cytometry and clonality testing may be helpful.
How is Mycosis Fungoides Treated?
Mycosis fungoides is treated depending on the stage of the skin lesions. For early skin lesions, steroids or UV light is used. For advanced skin lesions, aggressive systemic chemotherapy is indicated.
What is the Prognosis of Mycosis Fungoides?
The prognosis of mycosis fungoides is fair with a median survival for early stages of ≥ 20 years and median survival of late stages of the disease of around 5 years.
What is Sézary Syndrome?
Sézary syndrome is a leukemic variant of cutaneous T cell lymphoma defined by the presence of erythroderma, generalized lymphadenopathy and clonal T cells with cerebriform nuclei.
What is the Pathology of Sézary Syndrome?
The pathology of sezary syndrome is:
-Etiology: The cause of sezary syndrome is unknown.
-Genes involved: Unknown.
-Pathogenesis: The sequence of events that lead to sezary syndrome involves the expression of the adhesion molecule cutaneous leukocyte antigen and the chemokine receptors CCR4 and CCR10 which contribute to the homing of normal CD4+ T cells to the skin.
-Histology: The histology associated with sezary syndrome is similar with mycosis fungoides. Sezary cells are seen as atypical lymphocytes of intermediate to large size and cerebriform nuclei. Lymph node shows complete effacement by monotonous infiltration of the Sezary cells.
How does Sézary Syndrome Present?
Patients with sezary syndrome typically affects patients aged 60 to 70 with a male predilection of 2:1 ratio. The symptoms, features, and clinical findings associated with sezary syndrome include erythroderma as its key feature for diagnosis defined as involvement of >80% of body surface and may range from mild to intense. Palms and soles usually show thickening, scales and fissures. Alopecia and plaques may occur. Nail dystrophy, blepharoconjunctivitis and ectropium can be present in late stages of the disease. Systemic symptoms are usually absent.
How is Sézary Syndrome Diagnosed?
Sézary Syndrome is diagnosed based on clinical symptoms of erythroderma, generalized lymphadenopathy, clonal T cells and an absolute sezary cell count of ≥ 1 x 10^9/L on flow cytometry. An alternative diagnostic criteria on flow cytometry of ≥ 40% of CD4+ / CD7- and ≥ 30% of CD4+ / CD26- T cells may be helpful.
How is Sézary Syndrome Treated?
Sézary Syndrome is treated with systemic therapies of retinoids, bexarotene, recombinant interferon a, conventional chemotherapy (CHOP) are used. Target therapies of Alemtuzumab, anti-PDA1,CD47/SIRPa may also be used. In patients with chronic conditions that are incurable, mainstay of treatment is control of symptoms and improvement of quality of life.
What is the Prognosis of Sézary Syndrome?
The prognosis of sezary syndrome is poor with a median overall survival after diagnosis of 3.1-4 years. Median time to death after diagnosis is 3.5 years. 5 year overall survival range is 18-37%.
What is Anaplastic Large-Cell Lymphoma?
Anaplastic large-cell lymphoma is type of peripheral T cell lymphoma consisting of large lymphoid cells with abundant amphophillic cytoplasm, an ALK gene translocation and expression of ALK protein and CD30.
What is the Pathology of Anaplastic Large-Cell Lymphoma?
The pathology of anaplastic large-cell lymphoma is:
-Etiology: The cause of anaplastic large-cell lymphoma is the presence of rearrangements in the ALK gene on chromosome 2p23.
-Genes involved: ALK gene.
-Pathogenesis: The sequence of events that lead to anaplastic large-cell lymphoma involve the rearrangements in the ALK gene on chromosome 2p23 which break the ALK locus and lead to the formation of chimeric genes encoding ALK fusion proteins constitutively active tyrosine kinases that trigger the RAS and JAK/STAT signaling pathways.
-Histology: The histology associated with anaplastic large-cell lymphoma shows large anaplastic cells, some containing horseshoe shaped nuclei and voluminous cytoplasm. The tumor cells cluster about venules and infiltrate lymphoid sinuses mimicking the appearance of metastatic carcinoma.
How does Anaplastic Large-Cell Lymphoma Present?
Patients with anaplastic large-cell lymphoma typically affect children and young adults in the first 3 decades of life with a male predominance ratio of 1.5:1. The symptoms, features, and clinical findings associated with anaplastic large-cell lymphoma include fever, weight loss, night sweats, peripheral or abdominal lymphadenopathy, extranodal infiltrates, bone marrow involvement, anemia and pancytopenia.
How is Anaplastic Large-Cell Lymphoma Diagnosed?
Anaplastic Large-cell Lymphoma is diagnosed by biopsy of the lymph node or the tissue and examining its morphology along with immunohistochemistry. Immunophenotype and detection of genetic abnormalities and T cell clone can be helpful.
How is Anaplastic Large-Cell Lymphoma Treated?
Anaplastic Large-cell Lymphoma is treated with chemotherapy (CHOP treatment with or without Etoposide). Brentuximab vedotin and ALK inhibitors may be used for relapsed cases and for tumors that failed to respond to conventional chemotherapy.
What is the Prognosis of Anaplastic Large-Cell Lymphoma?
The prognosis of anaplastic large-cell lymphoma is very good in children and young adults because this frequently involves soft tissues. The cure rate with chemotherapy is 75-80%. In adults with ALK negative anaplastic large cell lymphoma, the prognosis is worse.
What is Angioimmunoblastic T-Cell Lymphoma?
Angioimmunoblastic T-cell lymphoma is a peripheral T cell lymphoma characterized by systemic disease, a polymorphous infiltrate involving lymph nodes and a prominent proliferation of high endothelial venules and follicular dendritic cells.
What is the Pathology of Angioimmunoblastic T-Cell Lymphoma?
The pathology of angioimmunoblastic t-cell lymphoma is:
-Etiology: The cause of angioimmunoblastic t-cell lymphoma is dysregulation of the follicular T helper cell leading to germinal center anarchy and development of the lymphoma.
-Genes involved: TET2, DNMT3A, IDH2, RHOA, CD28, PLCG1, TNFRSF21
-Pathogenesis: The sequence of events that lead to angioimmunoblastic t-cell lymphoma include an initial reaction of an unbalanced immune response to an unknown antigen followed by an oligoclonal phase driven by persistence and ineffective handling of the primary and initial stimulus
-Histology: The histology associated with angioimmunoblastic t-cell lymphoma shows partial effacement of lymphonodular architecture, perinodal infiltration with preservation of subcapsular and trabecular sinuses. There are also predominantly paracortical aggregates of polymorphic small to medium sized cells with clear/pale cytoplasm, distinct cell membranes and minimal cytologic atypia. Bone marrow involvement may contain polymorphous infiltrates of lymphocytes, neutrophils, histiocytes and eosinophils along with vascular proliferation with prominent endothelial cells and fibroblasts.
How does Angioimmunoblastic T-cell Lymphoma Present?
Patients with angioimmunoblastic t-cell lymphoma typically present with no gender predisposition with a median age of 65 years. The symptoms, features, and clinical findings associated with angioimmunoblastic t-cell lymphoma include fever, night sweats, weight loss, generalized lymphadenopathy, hepatosplenomegaly and skin rash. Polyclonal hypergammaglobulinemia and autoimmune hemolytic anemia may develop with progression.
How is Angioimmunoblastic T-cell Lymphoma Diagnosed?
Angioimmunoblastic T-cell Lymphoma is diagnosed mainly by excisional biopsy of an affected lymph node. Testing for clonal T cell receptor rearrangement may be helpful.
How is Angioimmunoblastic T-cell Lymphoma Treated?
Angioimmunoblastic T-cell Lymphoma is treated with anthracycline containing regimens. Consolidative autologous transplantation may be considered. Studies show that romidepsin, belinostat, brentuximab, vedotin and lenalidomide are effective in pretreated patients.
What is the Prognosis of Angioimmunoblastic T-Cell Lymphoma?
The prognosis of angioimmunoblastic t-cell lymphoma is fair because it has a moderately aggressive course with occasional spontaneous remissions with a median survival of 24 months. Five year overall and failure free survivals were 33% and 18% respectively.